完全なテストデータが利用可能に – Novogeneが PacBio Kinnex完全長 RNA サービスを開始
Rui LIU、Novogene AMEA プロダクト マネージャー
2024年6月10日
従来のバルク RNA-seq では、選択的スプライシングの複雑さにより、完全なアイソフォーム構造を解明することが困難になることがよくあります。ロングリード シーケンスと HiFi データ (ベース精度 > 99.9%) を活用する PacBio の Iso-Seq 法により、5′ 末端から 3′ ポリ A テールまでの完全長転写産物を直接キャプチャできるようになりました。この画期的な進歩により、より正確なアイソフォームの発見と複雑な構造解析が可能になります。
新しい Kinnex Full-Length RNA アプローチは、コストとシーケンス深度のバランスを最適化することでオリジナルの Iso-Seq 法を強化し、研究効率を大幅に向上させます。MAS-Seq 法を利用することで、このキットは 8 個の cDNA 分子をより長いフラグメントに連結し、スループットを 8 倍に高めます。PacBio Revio システムと組み合わせると、細胞あたりのデータ出力は、Sequel II システムの通常の Iso-Seq 法と比較して 16 倍という驚異的な増加を示します (図 1)。このスループットの大幅な向上により、トランスクリプトーム データへのアクセスが容易になるだけでなく、研究コストも削減されます。
Kinnex 完全長RNAアプローチは、転写産物のキャプチャーにおいて優れた感度を示します。飽和曲線で示されるように、10Mb HiFi リードでは、既知のアイソフォームの約 80% を検出できます (図 2)。これは、この量で既知のアイソフォームの大部分を検出するのに十分であることを示しています。シーケンス深度をさらに増やすと、より希少なアイソフォームを検出できるようになり、より包括的なトランスクリプトーム カバレッジが提供されます。
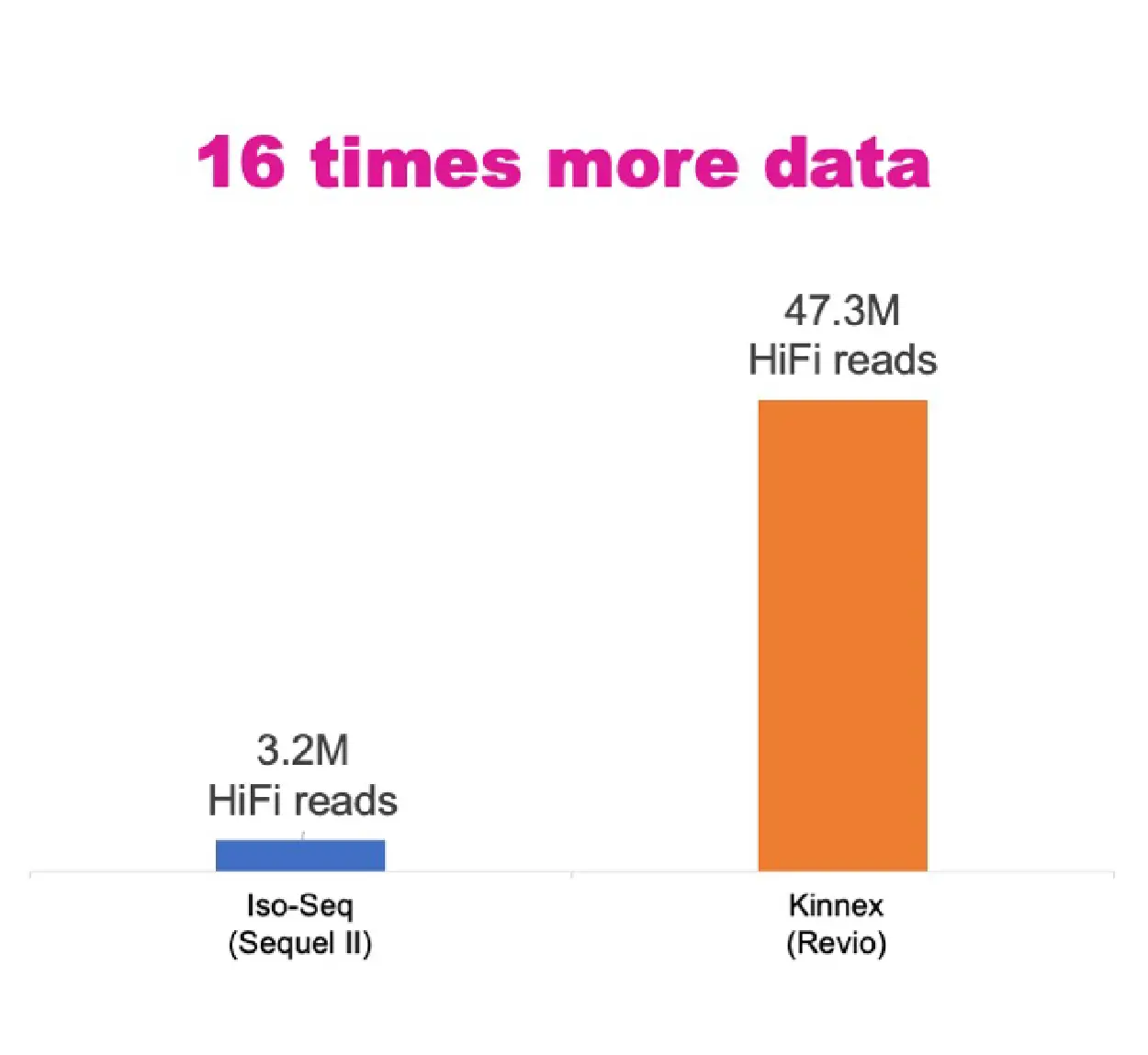
図1. ユニバーサルヒト参照RNA(UHRR)サンプルにおけるセル当たりの収量の比較:Sequel IIシステムでの通常のIso-SeqライブラリとRevioシステムでのKinnexライブラリ
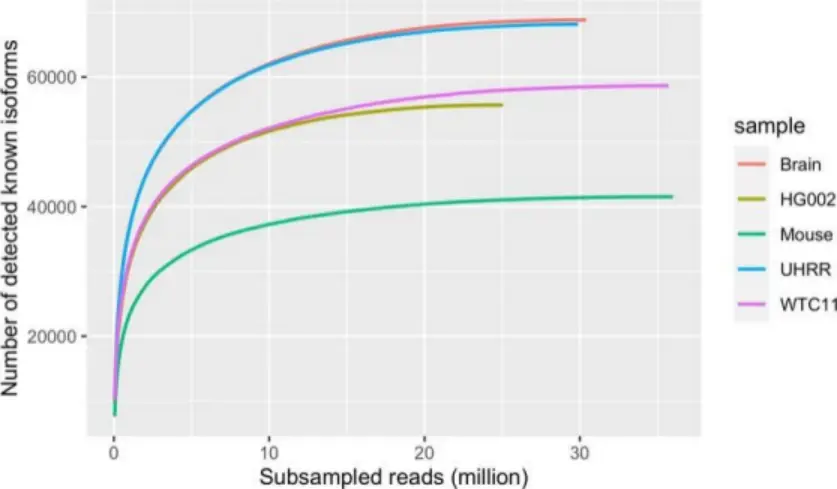
図2.異なるKinnexサンプルにおける既知のアイソフォームの飽和解析
1.1.収量能とデータ品質
1つのREVIO SMRTセルでシーケンスされた8プレックスのヒトライブラリから、合計39MbのHiFiリードそして各サンプルからは約5MのHiFiリードが得られ、データセット全体の均一性が非常に高いことが示されました(表1)。データは優れた品質を示し、平均リード品質指標はQ35を上回りました(図3)。
| Sample | HiFi Read Number | Read N50(bp) |
| Sample 1 | 4,973,898 | 2017 |
| Sample 2 | 5,046,454 | 1875 |
| Sample 3 | 4,799,817 | 1912 |
| Sample 4 | 4,320,335 | 1914 |
| Sample 5 | 4,839,172 | 1983 |
| Sample 6 | 4,920,919 | 1910 |
| Sample 7 | 5,073,452 | 1898 |
| Sample 8 | 5,058,747 | 1912 |
表1. データ出力とリード長の状態
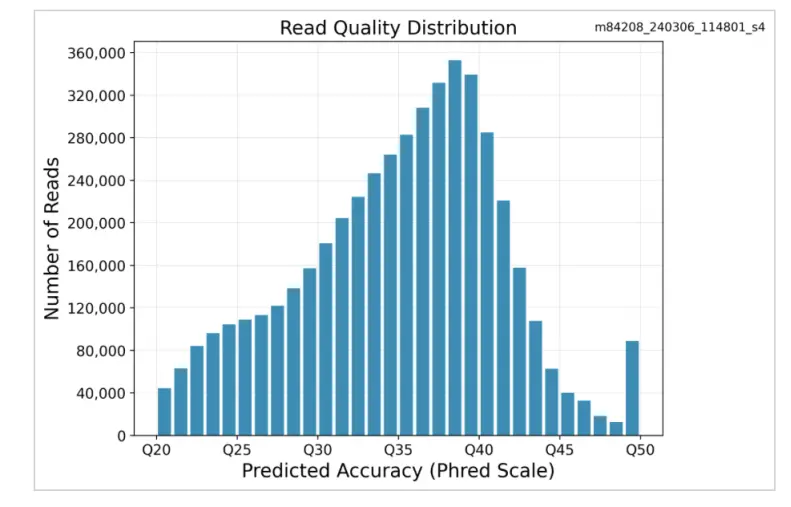
図3. リード品質分布
2.転写産物の構造解析
一つのヒト組織サンプルから、合計4.96Mbの全長非キメラ(FLNC)シーケンスが得られました。これらのシーケンスを転写産物の構造解析に利用しました。SQANTI32を用いて、合計106,012のユニークな全長転写産物を同定しました。注目すべきは、この研究で分類された転写産物の36%が新規転写産物(NIC+NNC)であったことです(図4)。
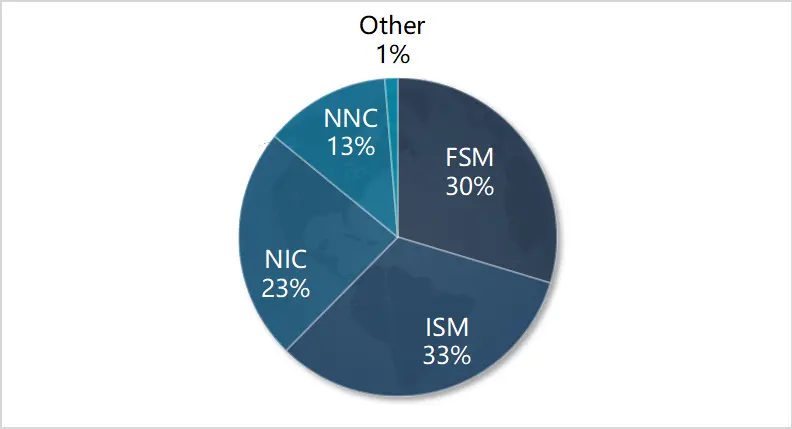

図4. 転写物の分類。注:FSM:全てのSJに完全に一致 ISM:参照SJに部分的に一致 NiC:既知のスプライス部位の新しい組み合わせを持つ新規アイソフォーム NNC:少なくとも新しいスプライス部位を持つ新規アイソフォーム
ノボジーンを選ぶ理由
1. 増加した納品データ量
ノボジーンでは、研究者の多様なニーズに合わせた新しい納品方法を導入しています。サンプルあたり5M HiFiリードと10M HiFiリードの2つのパッケージを提供し、HiFiデータを直接お届けします。
|
アプリケーション |
ターゲットデプス |
ターゲットデプス |
|
低から中程度発現している転写物のアイソフォーム発見と定量 |
サンプルあたり |
複数の繰り返しで腫瘍vs正常組織の比較 |
|
高発現している転写物のアイソフォーム発見 |
サンプルあたり5M read |
30サンプル以上の患者コホート研究 |
|
種における包括的な転写物アノテーション |
サンプルあたり5M read |
複数の組織タイプからの |
表2. Kinnex 全長RNAアプリケーションの使用例
2.強化された解析ワークフロー
ノボジーンでは、正確なアイソフォーム分類のためにSQANTI3ソフトウェアを利用し、遺伝子および転写産物の発現レベルの詳細な解析のためにIsoQuant3.33で補完しています。これら2つのソフトウェアの組み合わせにより、研究者はトランスクリプトームデータから貴重な知見を発見することができます。
SQANTI3ソフトウェアのハイライト:
- ロングリードRNAシーケンスデータ用に特別に設計され、転写産物の構造を特徴付けます。
- 転写開始および終了部位、スプライシングジャンクション、その他の構造的特徴など、転写産物の構造特性を特徴付ける複数の指標を組み込み、潜在的な偽陽性やその他のエラーをフィルタリングできます。
- 信頼性の高いアノテーションを優先し、解析中に重要な遺伝子や転写産物の情報を保持するRescueモジュールを含みます。新規転写産物を参照データベースと比較することにより、適切にアノテーションされた転写産物の保持を保証します。
参考文献
- Al’Khafaji AM, Smith JT, Garimella KV, Babadi M, Popic V, Sade-Feldman M, et al. High-throughput RNA isoform sequencing using programmed cDNA concatenation. Nat Biotechnol. 2023. doi: 10.1038/s41587-023-01815-7
- Pardo-Palacios FJ, Arzalluz-Luque A, Kondratova L, Salguero P, Mestre-Tomás J, Amorín R, et al. SQANTI3: curation of long-read transcriptomes for accurate identification of known and novel isoforms. Nat Methods. 2024 May 1;21(5):793-797. doi: 10.1038/s41592-024-02229-2.
- Prjibelski A, Pfeil R, Mikheenko A, Webber J, Tomescu A, Tilgner H. IsoQuant: A tool for isoform assignment and quantification for long and barcoded reads [Internet]. University of Helsinki and Saint Petersburg State University; 2022-2024. Available from: https://github.com/ablab/IsoQuant.