概要
PacBio SMRT(Single Molecule, Real-Time)技術を用いたアイソフォームシーケンス(Iso-Seq)は、標的遺伝子内の全長トランスクリプトアイソフォーム(5’UTRから3’ポリAテールまで)のシーケンスを可能にします。Iso-Seqは、融合遺伝子の特性解析、選択的スプライシングの同定、ゲノムアノテーション、新規転写産物の発見のためのハイスループットな手法です。
Iso-Seqは、疾患メカニズムの解明、薬剤耐性メカニズムの探索、新規遺伝子の発見、植物発生や生物的・非生物的ストレスの研究など、医療・農業研究分野で最大限に活用できます。

アイソフォームシーケンスのアプリケーション
- 疾患メカニズムの解明
- 潜在的バイオマーカーとしての選択的スプライシング転写産物の発見
- 薬剤耐性メカニズムの解明
- 新規遺伝子・転写産物の同定
- 遺伝子のコード領域、調節要素、構造要素を認識するためのゲノムアノテーションの改善
- 環境ストレス下における植物発生の理解
- 選択的スプライシングに起因する新規アイソフォームの発見
アイソフォームシーケンスの利点
- 業界をリードするシーケンス能力、高品質データ、迅速な納期、手頃な価格
- 新規転写産物の発見、変動発現解析、機能アノテーション解析を可能とする確立されたパイプライン
- リード長とデータ出力においてPacBioの基準を超えるシーケンスプロセスの最適化能力
Kinnex全長RNAキット
Kinnexキットは、スループット向上のために小さなアンプリコンを連結して大きな断片ライブラリを生成するMAS-Seq法に基づいています。ノボジーンは、さらにスループットを向上するようアイソフォームシーケンスパイプラインの一環としてKinnex全長RNAキットとPacBio Revioシステムを採用しています。これにより、ショートリードRNAシーケンスでは達成困難な解像度での大規模研究が可能となります。
アイソフォームシーケンス仕様:サンプル要件
| ライブラリタイプ | サンプルタイプ | サンプル量 | 濃度 | RIN (Agilent 2100) |
純度 (Nanodrop TM/Agarose Gel) |
| PacBio Kinnex全長RNAライブラリ | Total RNA | ≥ 600 ng | ≥ 40 ng/μL | ≥ 6.5 | A260/280= 1.8-2.2; A260/230= 1.3-2.5; *NC/QC≤2 |
アイソフォームシーケンス仕様:シーケンスと解析
| プラットフォーム | PacBio Revio |
| ライブラリタイプ | Kinnex 全長RNAライブラリ |
| 推奨データ量 | サンプル当たり 5M/10M HiFi reads |
| 解析内容 |
参照ゲノムあり
参照ゲノムなし Kinnex全長RNAライブラリーを用いた10M HiFiリードを選択した場合、ショートリードmRNA-seqを補助とせずとも定量解析が可能 (転写産物解析は参照ゲノムが利用可能な場合にのみ実施可能) |
アイソフォームシーケンスのプロジェクトワークフロー
ノボジーンは、プロジェクトワークフロー全体を通じて高品質な製品と専門的なサービスを提供します。各ステップは厳格な科学的基準を満たすよう慎重に設計・実行され、卓越した研究成果を保証します。シーケンスデータの正確性と信頼性を確保するため、プロセスの各段階で厳格な品質管理(QC)措置を実施しています。ワークフローには、サンプル調製・定量、ライブラリ調製、ライブラリ品質管理、シーケンス、バイオインフォマティクス解析などの主要ステップが含まれます。




注目の論文
-
Scientific&DataDate: January 2019IF: 5.929DOI: https://doi.org/10.1038/s41597-019-0240-1
CCS
各リードのCCS(Circular Consensus Sequence)は、単一のZMW(ゼロモード導波管)から得られたサブリードを相互に補正・アラインメントすることで作成できます。
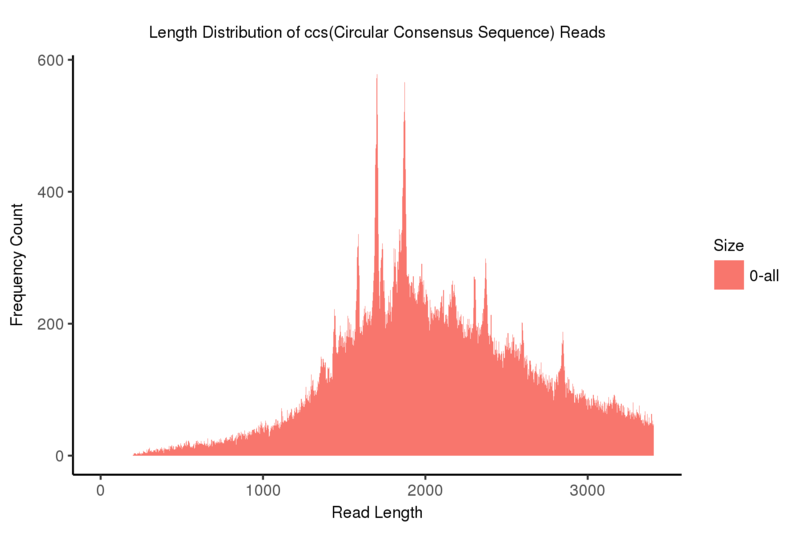
Length distribution of CCS(Circular Consensus Sequence) reads
注:
x軸はリード長、y軸はリード長に対応する頻度カウントを示します。
構造カテゴリー
特性評価結果に基づくアイソフォーム数の分布。NICまたはNNC(新規アイソフォーム)のアイソフォーム数が顕著に多くなっています(左);通常、特に新規遺伝子では、ほとんどの場合で一遺伝子一アイソフォームの分布が観察されます(右)
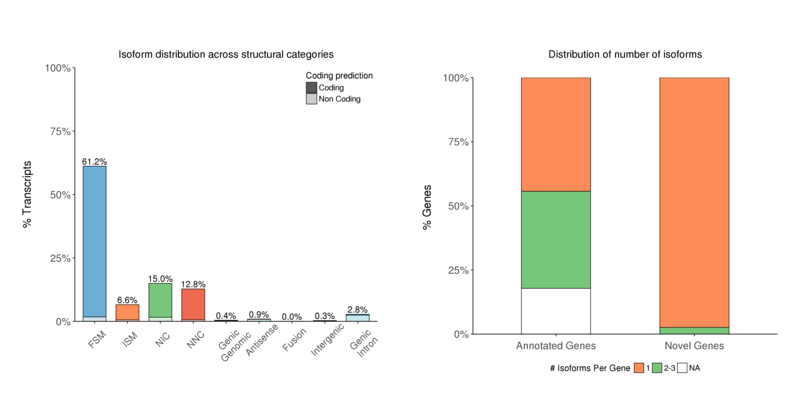
Isoform numbers by structural category (left) and by genetype (right)
注:
x軸:アイソフォーム分類 y軸:各分類におけるアイソフォーム割合(左図)
x軸:遺伝子タイプ y軸:「遺伝子あたりのアイソフォーム数」分類における遺伝子割合(右図)
長さの分布
構造分類に基づくアイソフォームの転写産物長分布とエクソン数分布を箱ひげ図で示しています。
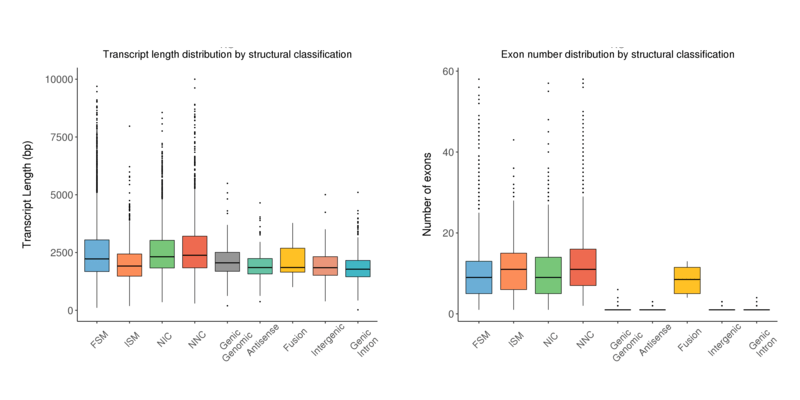
Transcript length distribution by structural classification (left) and exon numbers distribution by structural classification (right) by transcript type
注:
x軸は転写産物分類を示します;左y軸は各分類における転写産物の長さを示します;右y軸は各分類における転写産物のエクソン数を示します
構造と機能アノテーション
ジーンオントロジー
ジーンオントロジー(GO)プロジェクトは、複数のデータベース内で遺伝子産物を信頼性の高い記述で提供することを目的としています。GO語彙(オントロジー)は、生物種に依存しないアプローチで、遺伝子産物に関連する生物学的プロセス、分子機能、細胞構成要素について説明しています。GOアノテーションは、同定された新規遺伝子およびアイソフォームに対してのみ利用可能です。
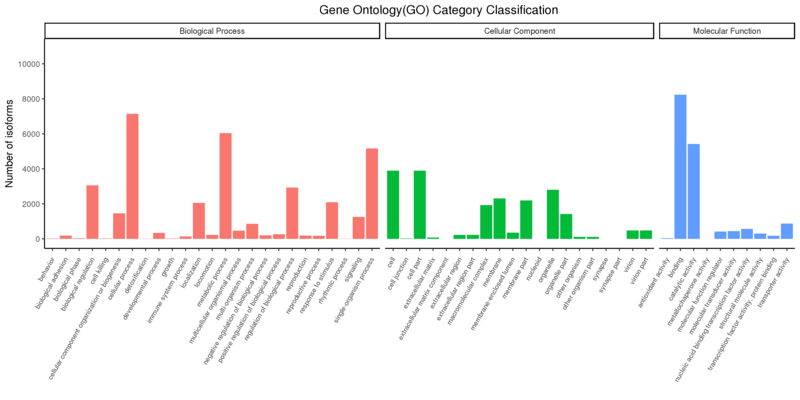
Gene Ontology Annotation Classification
注:
x軸は3つのGOカテゴリーを示し、y軸は当該ターム(そのサブタームを含む)にアノテーションされた変動発現遺伝子の数を示しています。3つの異なるカテゴリーはGOタームの3つの基本分類(左から生物学的プロセス、細胞構成要素、分子機能)を表しています
構造解析
CNCI(Coding-Non-Coding Index)は、配列の固有構成に基づく予測を行う強力なシグネチャツールであり、全トランスクリプトームシーケンスデータからアセンブルされた転写産物の正確な分類を提供します。PLEK(改良k-merスキームに基づくロングノンコーディングRNAおよびメッセンジャーRNA予測ツール)は、改良k-merスキームとサポートベクターマシン(SVM)アルゴリズムに基づく計算パイプラインを用い、ゲノム配列やアノテーションが欠如している状況下でロングノンコーディングRNAおよびmRNAを予測するツールです。PLEKとCNCIの結果をベン図で示します。
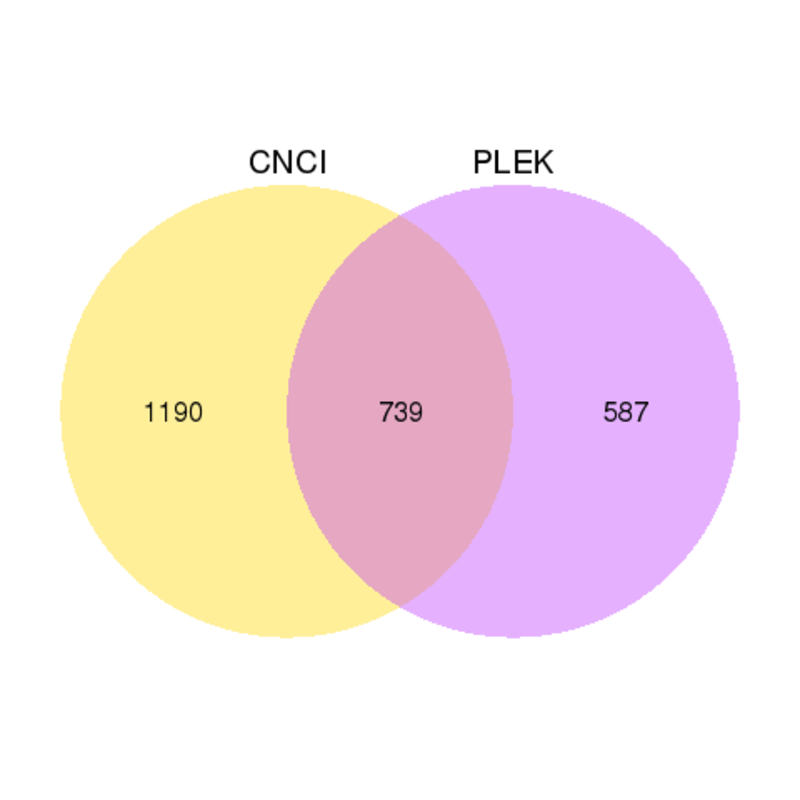
Venn diagrams of results from PLEK and CNCI
注:
x軸は3つのGOカテゴリーを示し、y軸は当該ターム(そのサブタームを含む)にアノテーションされた変動発現遺伝子の数を示しています。3つの異なるカテゴリーはGOタームの3つの基本分類(左から生物学的プロセス、細胞構成要素、分子機能)を表しています
選択的スプライシング
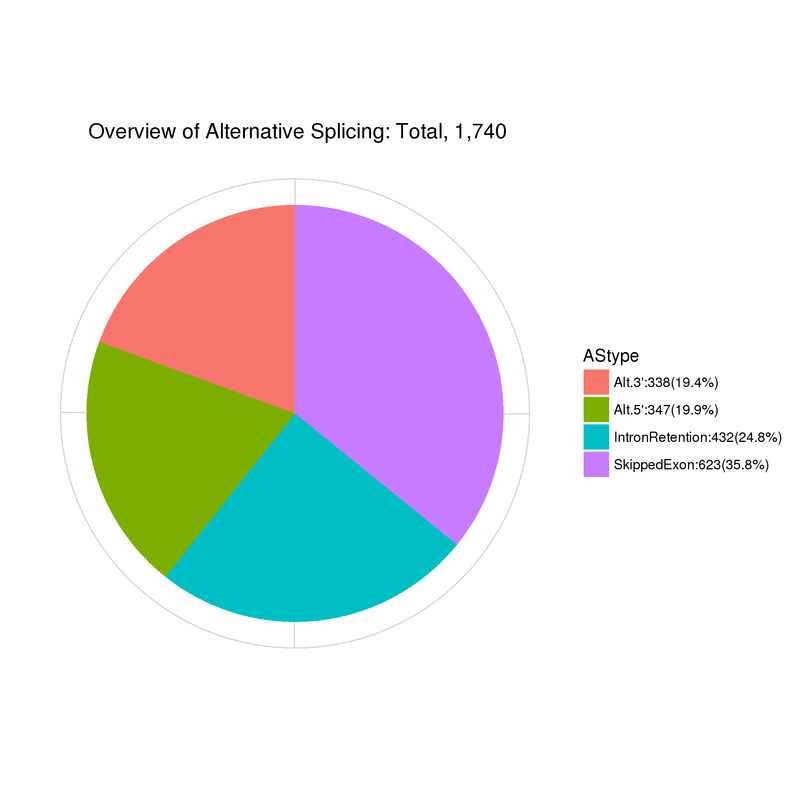
Gene Ontology Annotation Classification
注:
Alt.3’: 選択的5′スプライス部位; Alt.5’: 選択的3′スプライス部位
*完全版のデモレポートをご希望の場合は、お問い合わせください。