概要
ヒト全ゲノムシーケンス(hWGS)により、研究者は個々の全遺伝子構成をカタログ化し、ヒトゲノム全体を特徴付けることができます。一塩基多型(SNPs)、挿入および欠失(InDels)、構造変異(SVs)、コピー数多型(CNVs)などのゲノム変異情報を、単一のコスト効率の高いアッセイで同定することができます。
豊富な経験とバイオインフォマティクスのノウハウを生かし、ノボジーンは様々な研究目的やお客様のニーズにお応えするために、高品質なデータ、論文発表可能な解析結果、個別の結果をお届けします。ノボジーンは、大規模なプロジェクトにおいても迅速な納期で結果をご提供します。:多数のIllumina NovaSeq X Plus /NovaSeq6000プラットフォーム、Oxford Nanopore PromethION、PacBio Revio/Sequel IIシステムを備えています。
ノボジーンは、競争力のあるコストで年間最大20万ヒトゲノムのシーケンスが可能です。ノボジーンのヒトWGSサービスは、遺伝性疾患、がん、発症メカニズム、または集団遺伝学に関する研究を含む、幅広い用にデータを提供することができます。ノボジーンで利用可能な複数のDNAシーケンス技術は、目的のゲノム内の高度多型および高度反復領域を同定することができ、それにより完全かつ正確なヒトゲノムの特性解析を提供します[1]。

ヒト全ゲノムシーケンスのアプリケーション
ヒトゲノムシーケンスは、以下の研究分野で研究者を支援します:
- 遺伝性疾患
- 発症メカニズム
- がん
- ヒト集団の起源
利点
ショートリードシーケンス
- SNP、InDels、SV、CNVなどのゲノムバリアントを効率的に発見、報告。
- 大規模コホートや複雑な疾患研究で利用可能なゲノムワイド関連研究(GWAS)のための費用対効果の高い戦略、ローパスシーケンス、およびインピュテーションサービス。
ロングリードシーケンス
- 繰り返し配列や大規模な構造変異を効率的に発見。
- メチル化情報(CpG部位の5mC)の直接検出。
仕様:DNAサンプル要件
| プラットフォームタイプ | サンプルタイプ | サンプル量 (Qubit) | 純度 (NanoDrop™/Agarose Gel) |
|
Illumina
NovaSeq X Plus /NovaSeq6000 |
Genomic DNA | ≥ 200 ng |
A260/280=1.8-2.0;
no degradation,
no contamination |
| Genomic DNA (PCR free) |
≥ 1.2 μg | ||
| Genomic DNA from FFPE tissue |
≥ 400 ng | Fragments longer than 1,000 bp | |
| PacBio Revio/ Sequel II system HiFi library | HMW Genomic DNA | ≥5 μg | A260/280=1.75-2.0; A260/230=1.4-2.6; *NC/QC=0.95-3.00 Fragments should be ≥ 30 kb |
| Nanopore PromethION |
HMW Genomic DNA | ≥ 8.5 μg | A260/280=1.75-2.0; A260/230=1.4-2.6; Fragments should be ≥ 30 kb |
*NC/QC: NanoDrop concentration/Qubit concentration
シーケンスパラメーターと解析内容
| Platform Type | Illumina NovaSeq X Plus /NovaSeq6000 | PacBio Revio/ Sequel II system | Nanopore PromethION |
| Read Length | Paired-end 150 bp | > 15 kb (Average) |
> 17 kb (Average) |
|
Sequencing Depth
|
For rare diseases: 30-50× |
For genetic diseases: 10-20× |
For genetic diseases: 10-20× |
| For tumor tissues: 50×; For adjacent normal tissues and blood: 30× |
For tumor tissues: ≥20× |
For tumor tissues: ≥20× |
|
|
Standard Data
Analysis |
|
|
|
Note: Values of sequencing depths are only listed for your reference. For more information, please contact us.
プロジェクトワークフロー
サンプル調製、ライブラリー調製、DNAシーケンス、データQCからバイオインフォマティクス解析まで、ノボジーンは高品質なデータと専門的なサービスを提供します。各ステップは、高い科学水準と綿密な設計に基づき実施され、高品質な研究成果をお約束します。




データQC
シーケンスエラー率分布
シーケンスエラー率は、ディープシーケンスによる低頻度変異の正確な検出の主要な混乱を引き起こす因子です。これはシーケンスデータの品質を決定します。シーケンスエラー率は、シーケンスサイクルと大きく関連しており、イルミナのハイスループットシーケンスプラットフォームの一般的な特徴である試薬の消費により、各リードの終わりに向かって上昇します。
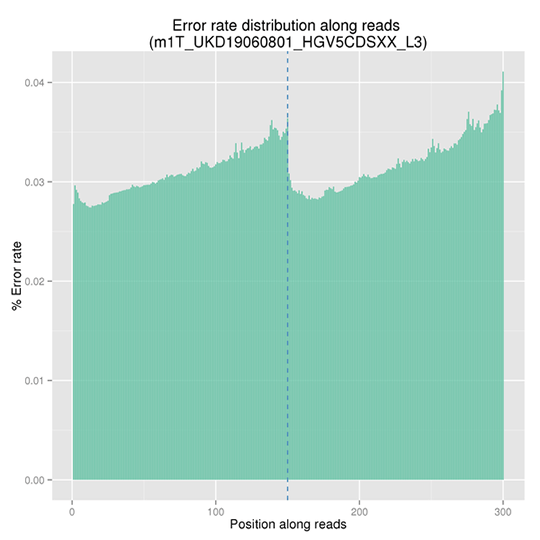
注:X軸はリードの位置を表し、Y軸はその位置における全リードの平均エラー率を表します。
GC含量分布
GC含量分布は、AT/GC分離の可能性をチェックすることを目的としています。サンプルのコンタミネーション、シーケンスバイアス、ライブラリー調製時のエラーは、シーケンス結果に影響を与える可能性があります。
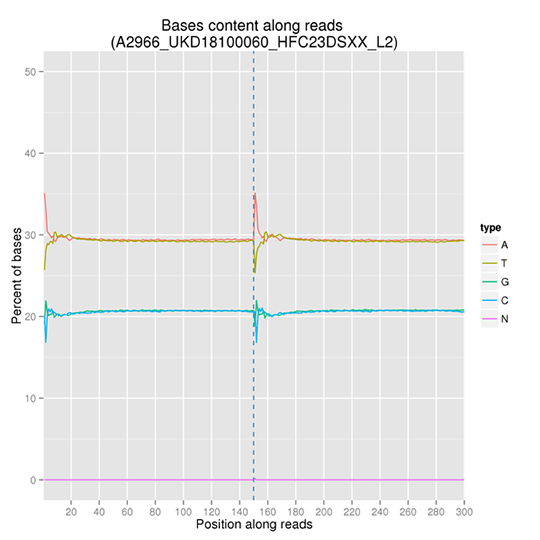
注:X軸はリード中の位置、Y軸は塩基の種類(A、T、G、C)ごとの割合で、異なる塩基は異なる色で区別できます。
リファレンスゲノムとのアラインメント
シーケンスデプスとカバレッジの分布
シーケンスデプスとカバレッジは、既知の参照ヌクレオチドにアライメントされたペアエンドクリーンリーンの平均数を示しています。シーケンスカバレッジ分布は、特定の塩基位置で変異の同定がある程度の信頼性をもって行えるかどうかを決定します。
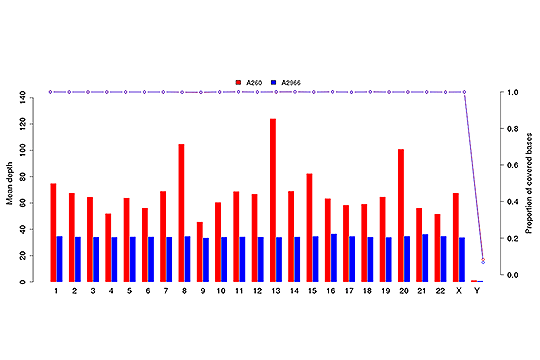
各染色体における平均シーケンスデプス(棒グラフ)とカバレッジ(点線グラフ)。
注:X軸は染色体を表し、左のY軸は平均デプス、右のY軸はカバレッジ(カバーされた塩基の割合)。
SNP/InDel/SV/CNVコール
一塩基多型(SNP)は一塩基変異(SNV)としても知られ、ゲノム中の遺伝的変異の最大のクラスを構成します。遺伝的変異の別のクラスには、長さが50bp未満の小さな挿入と欠失(InDels)が含まれる。コーディング領域やスプライシング部位に存在するInDelは、mRNA転写産物やタンパク質に変化を引き起こす可能性があります。
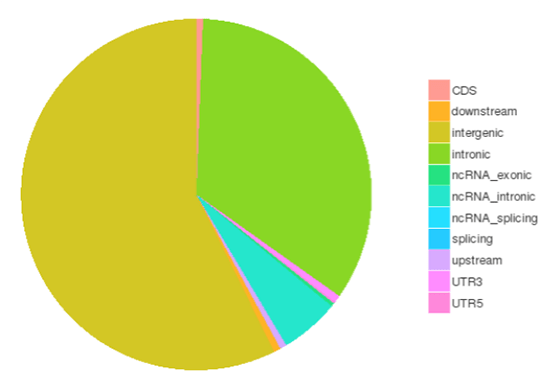
様々なゲノム領域におけるSNPs/InDelsの数
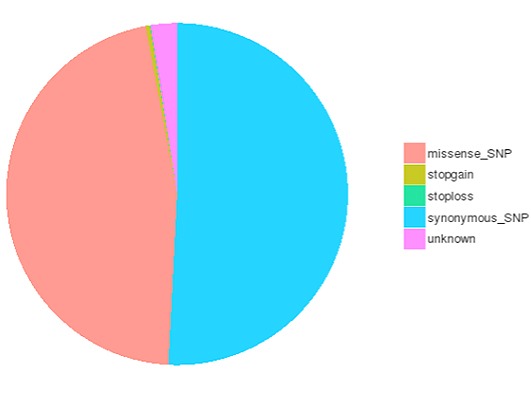
コーディング領域におけるSNP/InDelsの種類数
構造変異(SVs)は比較的大きなサイズ(>50 bp)の遺伝的変異で、欠失、重複、挿入、逆位、転座が含まれる。SVsは個人差の根底にある遺伝的基盤を形成し、疾患やがん感受性に影響を及ぼす可能性があります。
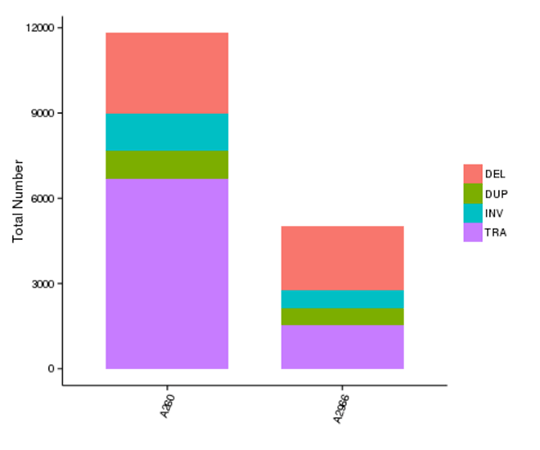
各サンプルにおける異なるタイプのSVの数
注:X軸はサンプルを示し、Y軸は各タイプのSVの数を示します。
コピー数変異体(CNV)とは、個体間で比較的大きな断片(50bpより長い)のコピー数が変動する遺伝的変異体である。CNVには2つのタイプ、すなわちコピー数の増加と減少がある。CNVは、個人差や癌の基礎となる遺伝的基盤を形成する可能性があります。
各サンプルにおけるCNVの影響を受けたゲノム領域の大きさ
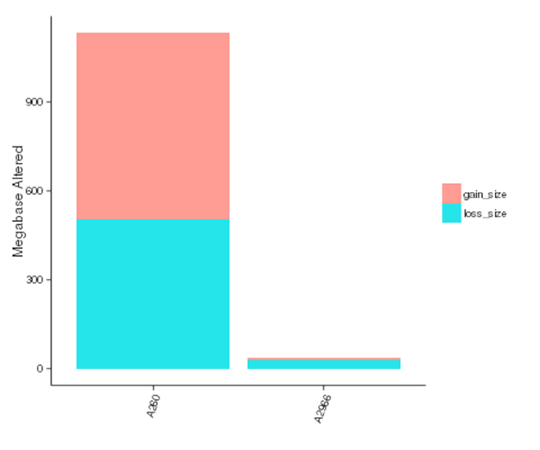
注:X軸はサンプル名、Y軸はゲノムの増減によって影響を受けた領域の合計サイズ(Mb)を示します。
高次解析
ドライバー遺伝子解析
有意に変異した遺伝子のヒートマップ
がんに関連する変異のうち、遺伝子に影響を与えることで腫瘍形成を促進するものはわずかである。有意に変異した遺伝子(SMGs)とは、バックグラウンド変異率(BMR)よりも有意に高い変異率を示す変異を指し、したがって腫瘍形成で陽性であることを示します。SMGの解析は、癌の発生と進行に重要な遺伝子を特定することに役立ちます。
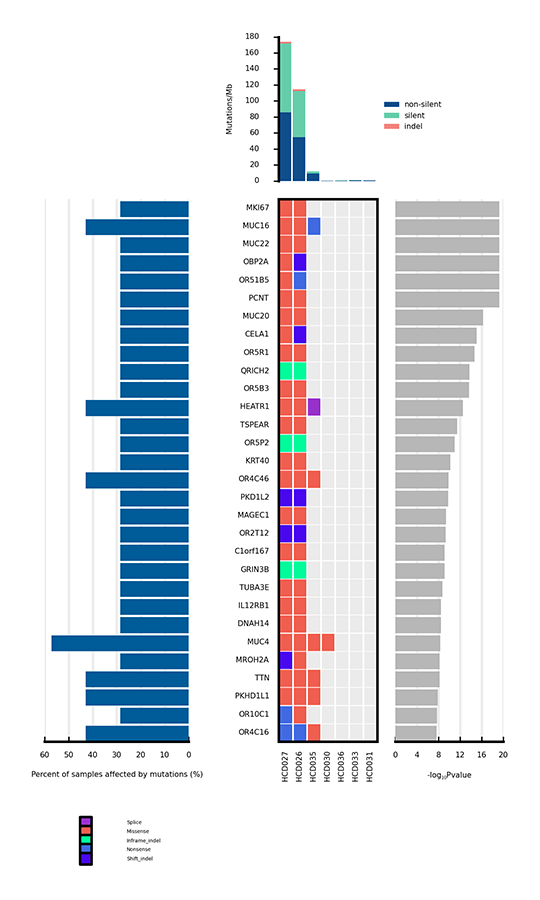
サンプル間で有意に変異した遺伝子(SMG)のヒートマップ
注
上部の棒グラフは各サンプルの変異率(Mutation/Mb)を示します。中央のヒートマップは、サンプル間の各SMGの変異の種類を示しています。横軸はサンプル、縦軸はSMGを表しています。異なるタイプの変異は異なる色で区別されています。ヒートマップの左側の棒グラフは、各SMGの変異の影響を受けたサンプルの割合を示し、右側のプロットはSMGのp値を示しています。
腫瘍不均一性解析
腫瘍内不均一性解析
腫瘍内不均一性とは腫瘍細胞の不均一な構成を指します。腫瘍内の不均一性とクローン構造を解読することは、治療抵抗性の理解に貢献する可能性があります。
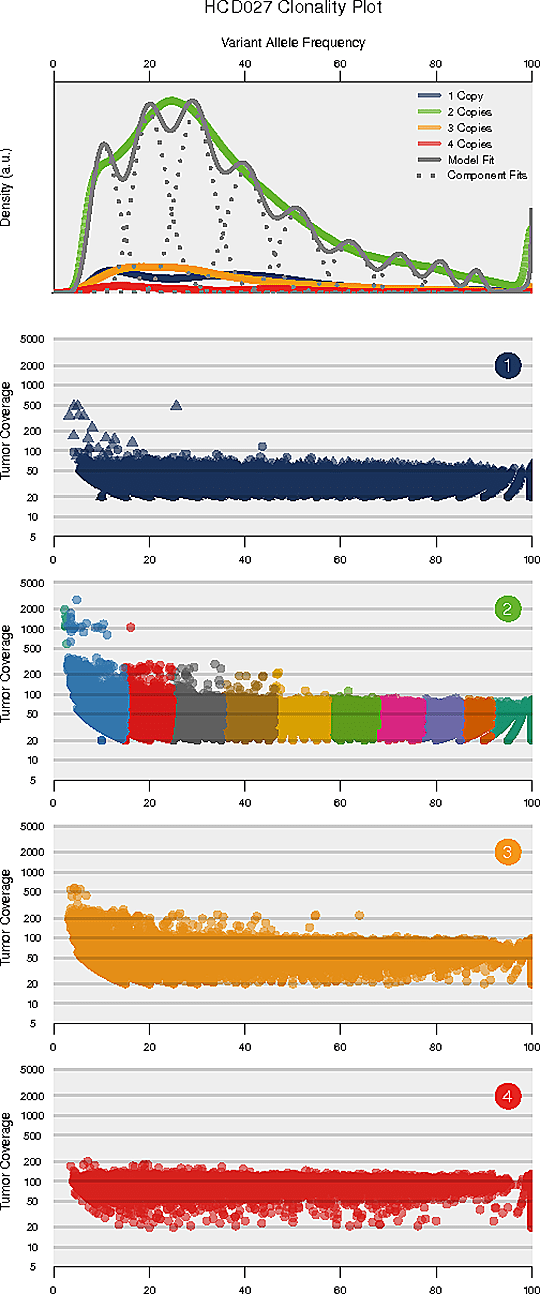
推定されるクローン構造
注
各パネルの横軸は変異対立遺伝子頻度(VAF)です。VAFが比較的低い変異のクラスターはサブクローン集団を表しています。上のパネルは、コピー数が1、2、3の領域全体のVAFのカーネル密度、コピー数が中立のバリアントについての全クラスターを合計した事後予測密度、各クラスター/構成要素の事後密度を示していなす。トップパネルの下のパネルは、コピー数領域の各クラスについて、リードデプスとVAFの関係を示しています。
*フルバージョンのデモレポートをご希望の方はお問い合わせください。