エクソームライブラリデータにおいて96%以上のSNP一致率、およびトランスクリプトームライブラリデータにおける97%以上の定量相関 NovaSeq X Plus 新データリリース
NovaSeq X Plusプラットフォームは、全ゲノムシ ケンスのみならず、全エクソームシ ケンスやトランスクリプトームシ ケンスにも並外れたシ ケンスパワ を発揮します。前回のNovaSeq X Plusでのヒト全ゲノムシ ケンステストのリリースに続き、より多くのライブラリタイプのデータを提示いたします。
1. ヒト全エクソームシーケンスライブラリの評価結
プラットフォームの性能をさらに評価するため、NA12878を含む全エクソームシ ケンスライブラリをNovaSeq X PlusとNovaSeq 6000の両方のプラットフォームで同時にシ ケンスを実施しました。ライブラリは、Agilent SureSelect Human All Exon V6を用いて調製しました。
1.1 QC結果
NovaSeq 6000と比較して、NovaSeq X PlusプラットフォームはよりQ30が高く、平均95.70%でした。
| Sample | Platform | Effective (%) | Error (%) | Q20 (%) | Q30 (%) | GC (%) | Containing N (%) | Low quality (%) | Adapter related (%) |
| S_W | NovaSeq 6000 | 97.88 | 0.02 | 98.31 | 95.19 | 51.97 | 0.00 | 0.00 | 2.11 |
| S_FC1_WL1X | NovaSeq X Plus | 97.19 | 0.03 | 97.59 | 95.73 | 51.26 | 0.01 | 0.00 | 2.8 |
| S_FC1_WL2X | NovaSeq X Plus | 97.00 | 0.02 | 97.87 | 96.13 | 51.26 | 0.01 | 0.00 | 2.99 |
| S_FC2_WL1X | NovaSeq X Plus | 97.83 | 0.03 | 97.23 | 95.56 | 50.58 | 0.00 | 0.00 | 2.17 |
| S_FC2_WL2X | NovaSeq X Plus | 97.73 | 0.03 | 97.21 | 95.37 | 50.66 | 0.00 | 0.00 | 2.27 |
1.2 マッピング統計値
NovaSeq X Plusは、エクソーム全体にわたって均一なカバレッジを達成し、カバレッジは99.5%、キャプチャ 効率は67.6%です。
| Sample | Platform | Mapped (%) | Properly (%) | PE (%) | Coverage (%) | 4X (%) | 10X (%) | 20X (%) |
| S_W | NovaSeq 6000 | 99.94 | 98.98 | 99.90 | 99.50 | 99.10 | 98.00 | 95.10 |
| S_FC1_WL1X | NovaSeq X Plus | 99.92 | 99.16 | 99.89 | 99.50 | 99.00 | 97.70 | 94.70 |
| S_FC1_WL2X | NovaSeq X Plus | 99.91 | 99.16 | 99.88 | 99.50 | 99.00 | 97.70 | 94.60 |
| S_FC2_WL1X | NovaSeq X Plus | 99.92 | 99.15 | 99.89 | 99.50 | 99.00 | 97.70 | 94.70 |
| S_FC2_WL2X | NovaSeq X Plus | 99.91 | 99.13 | 99.87 | 99.50 | 99.00 | 97.70 | 94.70 |
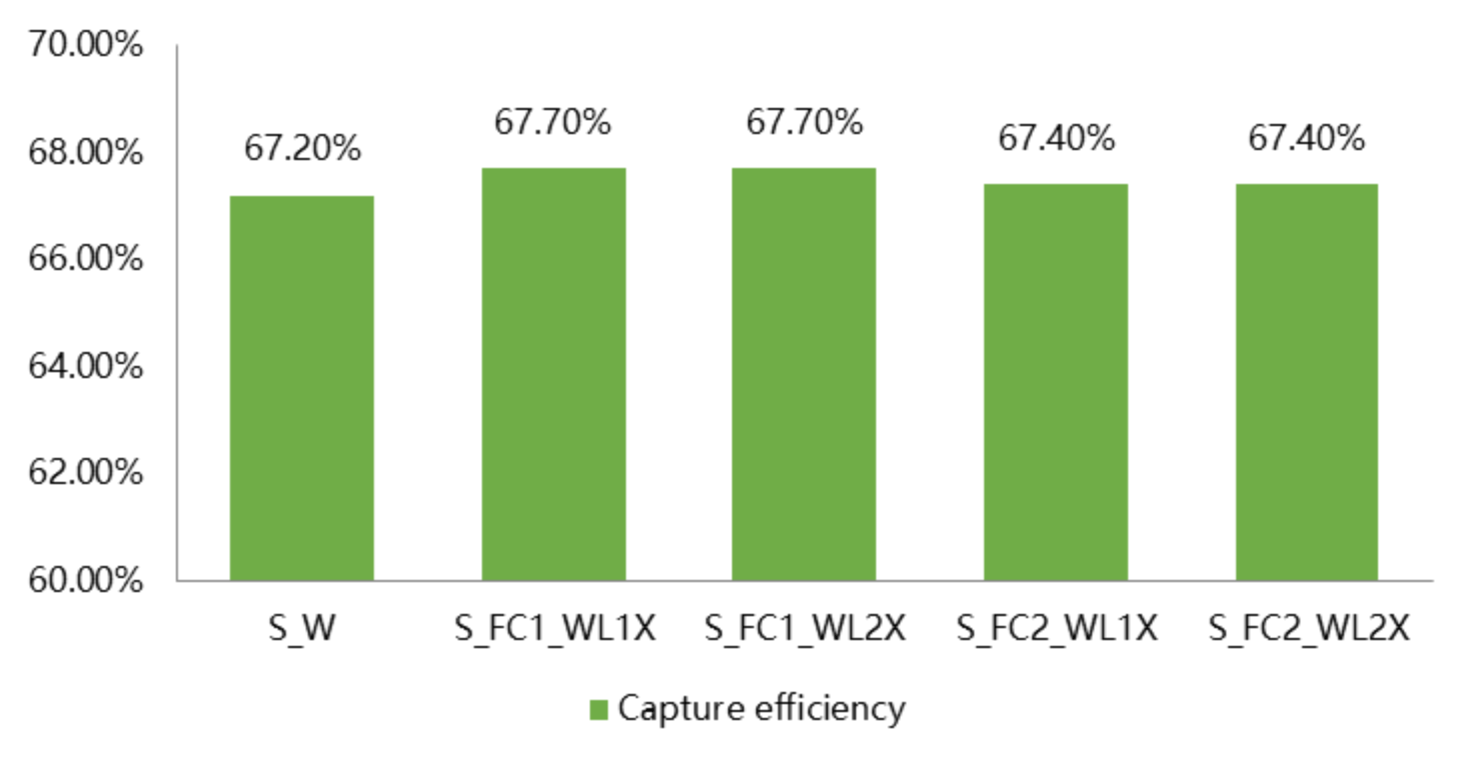
1.3 バリアントコ ル精度
NovaSeq X PlusとNovaSeq 6000プラットフォームで検出したSNPは96%以上が一致します。2つのプラットフォーム間では、SNPとInDelの適合率および再現率の偏差は、0.5%未満です。
| Sample | Platform | dbSNP (SNP) (%) | dbSNP (InDel)(%) | Precision (SNP) (%) | Recall (SNP) (%) | F-score (SNP) (%) | Precision (InDel) (%) | Recall (InDel) (%) | F-score (InDel) (%) |
| S_W | NovaSeq 6000 | 94.02 | 86.36 | 99.05 | 96.81 | 97.92 | 89.43 | 89.28 | 89.35 |
| S_FC1_WL1X | NovaSeq X Plus | 94.34 | 86.95 | 98.92 | 96.67 | 97.79 | 88.81 | 89.62 | 89.21 |
| S_FC1_WL2X | NovaSeq X Plus | 93.94 | 86.39 | 98.87 | 96.71 | 97.78 | 88.78 | 90.39 | 89.58 |
| S_FC2_WL1X | NovaSeq X Plus | 94.72 | 87.70 | 98.90 | 96.48 | 97.67 | 88.86 | 89.19 | 89.03 |
| S_FC2_WL2X | NovaSeq X Plus | 94.69 | 87.34 | 98.81 | 96.56 | 97.67 | 88.58 | 89.39 | 88.98 |
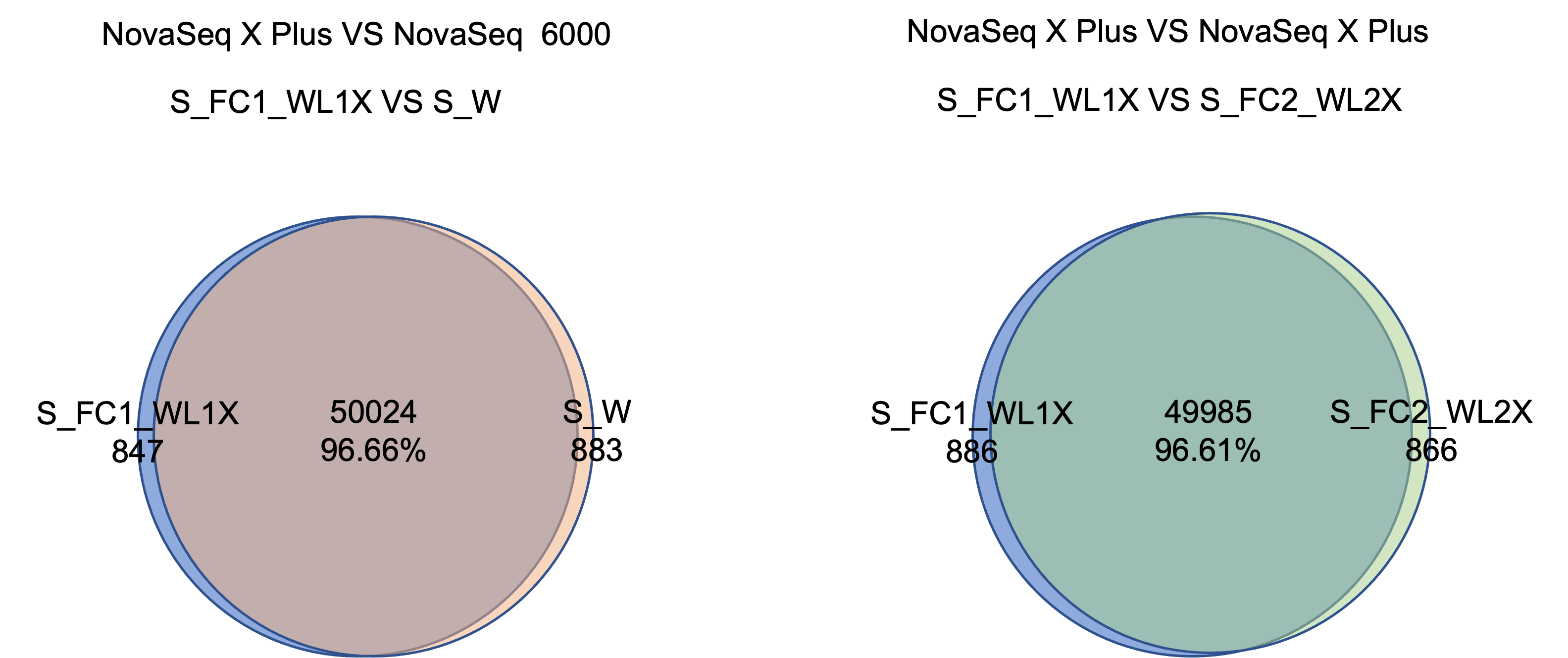
2. mRNAシーケンスライブラリの評価結果
UHRR (universal human reference RNA)を含むmRNAシ ケンスライブラリをNovaSeq X PlusとNovaSeq 6000の両方で同時にシ ケンスし、プラットフォームの性能をさらに評価しました。
2.1 QC結果およびマッピング統計値
NovaSeq X Plusの有効率、Q30スコア、rRNA率は、NovaSeq 6000とほぼ同等であり、偏差は1%未満です。
| Sample | Platform | Effective (%) | Error rate (%) | Q20 (%) | Q30 (%) | GC (%) | rRNA (%) |
| UHRR | NovaSeq 6000 | 95.85 | 0.02 | 98.28 | 95.05 | 50.15 | 2.24 |
| UHRR_L1 | NovaSeq X Plus | 94.21 | 0.03 | 97.33 | 95.28 | 49.19 | 1.48 |
| UHRR_L2 | NovaSeq X Plus | 93.85 | 0.02 | 97.70 | 95.79 | 49.15 | 1.24 |
| UHRR_L3 | NovaSeq X Plus | 95.54 | 0.03 | 97.07 | 95.25 | 48.47 | 1.58 |
| UHRR_L4 | NovaSeq X Plus | 95.44 | 0.03 | 97.02 | 95.02 | 48.55 | 1.23 |
| Sample | Platform | Mapped (%) | Unique map (%) | Multi map (%) | Exon (%) |
| UHRR | NovaSeq 6000 | 95.71 | 92.85 | 2.86 | 90.16 |
| UHRR_L1 | NovaSeq X Plus | 94.90 | 92.34 | 2.56 | 90.16 |
| UHRR_L2 | NovaSeq X Plus | 95.05 | 92.39 | 2.66 | 90.13 |
| UHRR_L3 | NovaSeq X Plus | 90.19 | 87.90 | 2.28 | 90.07 |
| UHRR_L4 | NovaSeq X Plus | 92.37 | 90.08 | 2.29 | 90.08 |
2.2 定量と相関
定量結果から、NovaSeq X PlusとNovaSeq 6000プラットフォームには、遺伝子発現に高い相似があることがわかりました。2つのプラットフォーム間のR2値は0.97以上であり、これはNovaSeq X Plusの2レーン間のR2値に匹敵します。
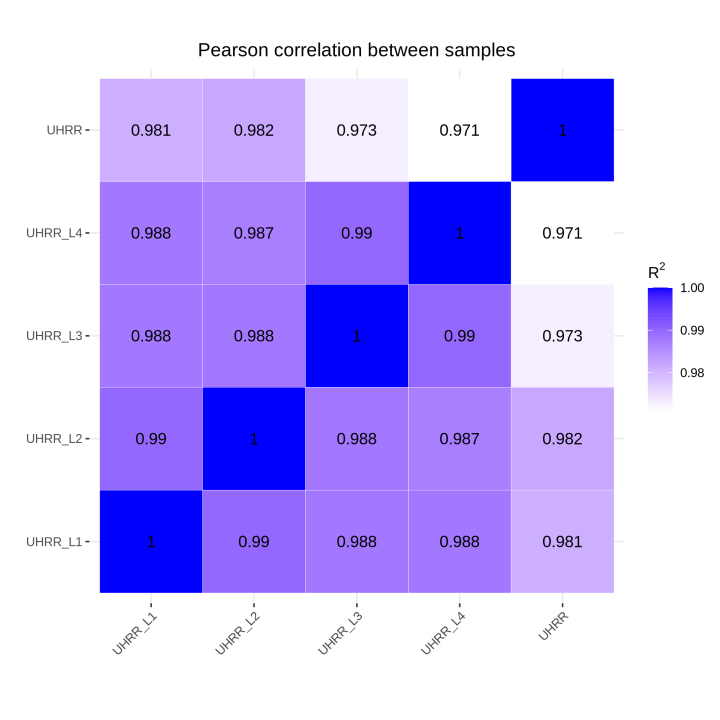
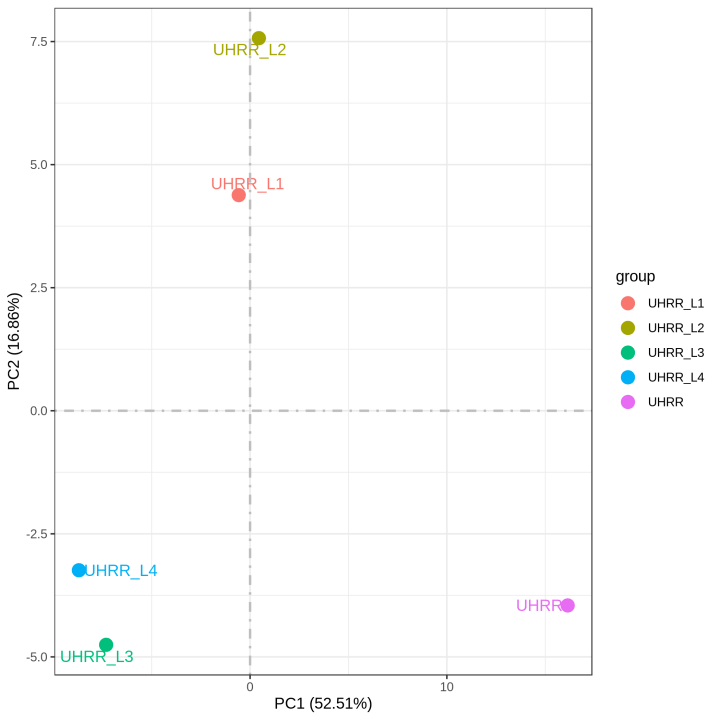
3. LncRNAシーケンスライブラリの評価結果
UHRR(universal human reference RNA)を含むLncRNAシ ケンスライブラリをNovaSeq X PlusとNovaSeq 6000で同時にシ ケンスし、プラットフォーム性能をさらに評価しました。
3.1 QCおよびマッピング統計値
NovaSeq 6000と比較すると、NovaSeq X PlusでのLncRNAシ ケンスライブラリの有効率、Q30、rRNA率はほぼ同じ結果を示し、偏差は1%未満でした。
| Sample | Platform | Effective (%) | Error rate (%) | Q20 (%) | Q30 (%) | GC (%) | rRNA (%) |
| UHRR_control | NovaSeq 6000 | 98.42 | 0.02 | 98.14 | 94.63 | 51.17 | 0.60 |
| UHRR_Lnc1 | NovaSeq X Plus | 97.74 | 0.03 | 97.09 | 94.83 | 49.94 | 0.29 |
| UHRR_Lnc2 | NovaSeq X Plus | 97.58 | 0.03 | 97.49 | 95.37 | 49.93 | 0.33 |
| UHRR_Lnc3 | NovaSeq X Plus | 98.16 | 0.03 | 96.83 | 94.63 | 49.20 | 0.27 |
| Sample | Platform | Mapped (%) | Unique map (%) | Multi map(%) | Exon map (%) |
| UHRR_control | NovaSeq 6000 | 96.83 | 93.54 | 3.29 | 68.14 |
| UHRR_Lnc1 | NovaSeq X Plus | 96.07 | 93.06 | 3.01 | 67.20 |
| UHRR_Lnc2 | NovaSeq X Plus | 96.22 | 93.09 | 3.12 | 67.11 |
| UHRR_Lnc3 | NovaSeq X Plus | 94.08 | 91.48 | 2.60 | 66.45 |
3.2 定量と相関
定量の結果、NovaSeq X PlusとNovaSeq 6000の遺伝子発現相似は高く、R2値は0.96以上でで、NovaSeq X Plusプラットフォームの2レーン間のR2値と同等であることがわかりました。
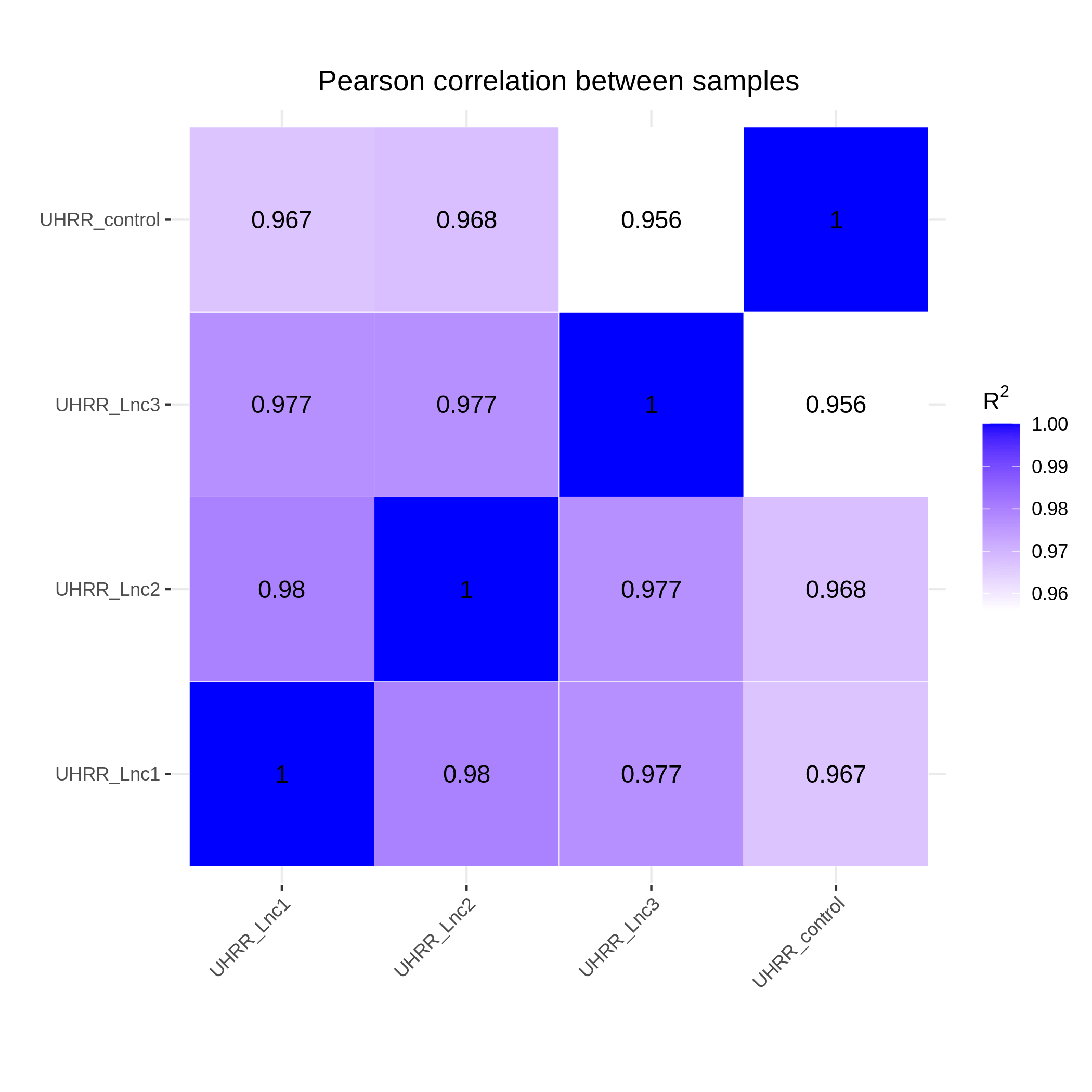
要約すると、NovaSeq X Plusプラットフォームは、全ゲノム、全エクソーム、トランスクリプトームシ ケンスライブラリにおいて、品質の向上およびNovaSeq 6000との高い 致を示しています。データ品質における優れた性能、納期、手頃な価格で、NovaSeq X Plusは集団ゲノム解析、遺伝子機能および制御に関連する研究にとって魅力的な選択肢となることでしょう。
ノボジーンについて
ノボジーンは、最先端の分子生物学技術とハイパフォ マンスコンピューティングを、ライフサイエンスとヒュ マンヘルスの分野の研究に応用するパイオニアです。そのビジョンは、ゲノミクスサ ビスとソリュ ションの提供において、グロ バルリーダ であり続けることです。
世界最大級のシ ケンスキャパシティを擁し、深い科学的知識、 流のカスタマ サ ビス、卓越したデータ品質を駆使して、急速に進化するゲノミクスの世界でお客様の研究目標を実現するお手伝いをします。ノボジーンは、お客様の信頼できるゲノミクスパートナ となることをお約束します。
(2) Error:エラー: read1とread2の全塩基の平均エラー率。塩基のエラー率は式1より求められる。
(3) Q20: phred-scaled quality scoreが20より大きい塩基の割合。
(4) Q30:Fred Scaleにて計算された品質スコアが30を超える塩基の割合。
(5) GC (%): 全塩基に含まれるGとCの割合。
(6) Containing N(%):どちらか一方のリードで、塩基が不確かなものが10%以上あるリードペア。
(7) Low Quality:どちらか一方のリードにおいて、低品質塩基の割合が50%以上であるリードペア。
(8) Adapter Related: どちらかのリードにアダプターの混入があるリードペア。
(9) Mapped (%): 参照ゲノムにマッピングされたリードの数(割合)。
(10) Properly: 適切にマッピングされたリードの数: 参照ゲノムにマッピングされたリードのうち、予想されるインサートサイズ内にあるリードの数(%)。
(11) PE mapped: 参照ゲノムにマッピングされたペアエンドリードの数(%)。
(12) Coverage: 全ゲノムにおけるカバレッジ。
(13) 4X: 4Xを超えるシーケンス深度の塩基のみを考慮した場合の全ゲノムにおけるカバー率。
(14) 10X (%):シーケンス深度10倍以上の塩基のみを考慮した場合の全ゲノムのカバー率。
(15) 20X (%):シーケンス深度20倍以上の塩基のみを考慮した場合の全ゲノムのカバー率。
(16) Precision=TP/(TP+FP) (パーセント)
(17) Recall=TP/(TP+FN)(パーセント)
(18) F score=2* Precision*Recall/(Precision + Recall)
(19) dbSNP(SNP).dbSNPデータベースで報告されたSNPの数をSNPの総数で割ったものです。
(20) dbSNP (InDel): dbSNPデータベースで報告されているInDelの数をInDelの総数で割った値。
(21) Unique_map: 参照ゲノムのユニークポジションにアライメントされたリードの数と割合(その後の定量データ解析用)、ユニークマッピング率:(ユニークマップリード)/(トータルリード)*100。
(22) Multi_map:参照ゲノムの複数の位置にアライメントされたリードの数と割合、多重マッピング率:(多重マッピングリード)/(全リード)*100。
(23) Error_rate:平均シーケンスエラー率、Qphred=-10log10(e)で算出される。
(24) Exon:エクソン: ゲノムのエクソン領域にアライメントされたリードの数、およびクリーンリードに占める割合。
(25) Exon map: ゲノムのエクソン領域にアライメントされた塩基の数と、ゲノムにアライメントされた塩基数に対する割合です。