概要
A complete and accurate genome sequence is essential to the genomics study of new species and the investigation of complex structural genomic changes in wild relatives compared to published cultivar genome sequences. With de novo sequencing, the first genome map for a species is generated, providing a valuable reference sequence for phylogenetic studies, analysis of species diversity, mapping of specific traits and genetic markers, and other genomics research.
Service SpecificationsApplications
- Phylogenetic studies
- Evolution history
Advantages
- Highly experienced: We have completed major de novo genome sequencing projects, and our data has been published in top-tier journals.
- Bioinformatics expertise: Best-in-class and widely recognized software, such as Falcon and Canu, used for comprehensive plant and animal bioinformatic analysis.
- Diverse strategies: Incorporating sequencing results from various platforms including Illumina Novaseq, PacBio Sequel, and Oxford PromethION, we offer the best assembly solution tailored towards each unique genome.
Sample Requirements
| Platform Type | Sample Type | Amount (Qubit®) | Purity |
| Illumina Novaseq 6000 | Genomic DNA | ≥ 200 ng | OD260/280=1.8-2.0 |
| Genomic DNA (PCR free) | ≥ 1.5 μg | ||
| Genomic DNA from FFPE | ≥ 0.8 μg | ||
| PacBio Sequel I/II | HMW Genomic DNA | ≥ 10 μg (for Sequel I) ≥ 30 μg (for Sequel II) |
OD260/280=1.8-2.0; OD260/230=2.0-2.2; Fragments should be ≥ 30 Kb for Sequel I, ≥ 60 Kb for Sequel II |
| Nanopore PromethION | HMW Genomic DNA | ≥ 10 μg | OD260/280=1.8-2.0; OD260/230=2.0-2.2; Fragments should be ≥ 30 Kb |
Sequencing Parameters And Analysis Contents
| Sequencing Parameters | Illumina Novaseq 6000 | PacBio Sequel I/II | Nanopore PromethION | PacBio Revio |
| Read Length | Paired-end 150 bp | |||
| Recommended Sequencing Depth | For genome survey or assembly polishing: ≥ 50× | For genome assembly: ≥ 50× | For genome assembly: ≥ 30× | |
|
Standard Analysis
|
K-mer analysis | Long-read assembly | Long-read assembly | |
|
Assembly Statistics | Assembly Statistics | ||
|
Gene completeness evaluation | Gene completeness evaluation | ||
| Genome Annotation | - |
|
|
|
Note: For detailed information, please refer to the Service Specifications and contact us for customized requests.
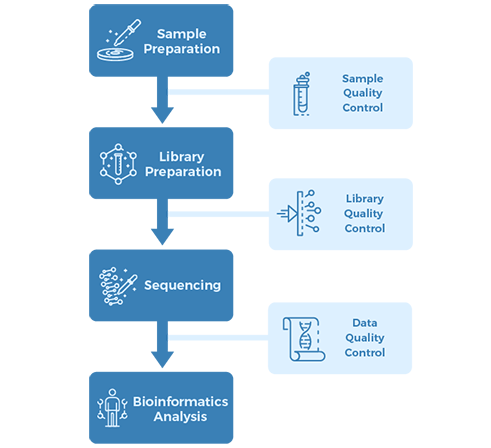
Project Workflow
Sampling & Sequencing Strategy:
Sampling:
• Fresh leaf tissues from single-living black pepper plants
• Library preparation: 350bp insert DNA library
• 20kb PacBio DNA SMRTbell library
• 10X Genomics DNA library
• BioNano DLS optical map
Sequencing: Illumina platforms and PacBio Sequel System
Results & Conclusion
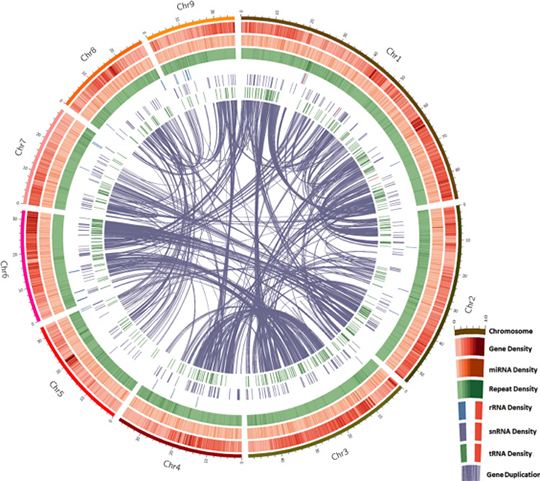
The 761.2 Mb sequences (45 scaffolds with an N50 of 29.8 Mb) are assembled into 26 pseudochromosomes (Figure. 1). This study therefore provides valuable insights that may serve as a foundation for future research on Piperales taxonomy and piperine biosynthesis, leading to a better understanding of the evolution, phytochemistry and ecology of the Piper genus.
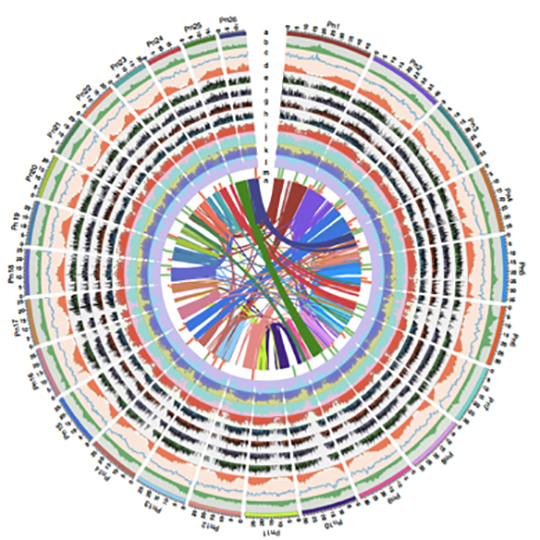
Chromosome‐level genome assembly of the razor clam Sinonovacula constricta (Lamarck, 1818)
Background:
Bivalves are highly diverse and the most evolutionarily successful class of invertebrates native to aquatic habitats. They provide valuable molecular resources for understanding evolutionary adaptation and aquatic ecology. The razor clam, Sinonovacula constricta (Lamarck, 1818) (family Solenidae, order Eulamellibranchia), is an economically, nutritionally, and ecologically important bivalve species native to the western Pacific coast. It is a representative organism of most benthic bivalves, but its genome has not been determined.
Sampling & Sequencing Strategy:
Sample:
• Wild S. constricta sampled from the natural mudflats
• Library preparation:
• PacBio SMRTbell DNA library
• Illumina short insert DNA library
• 10X Genomics DNA library
• Hi-C library
Sequencing: Illumina platforms and PacBio Sequel System
Results & Conclusion
A high-quality chromosome-level genome assembly of S. constricta was obtained with a total length of 1,220.85 Mb (Figure.2 Contig N50 of 976.94 Kb and a scaffold N50 of 65.93 Mb). The genomic resources provide a valuable reference genome not only for S. constricta’s genetic improvement and disease treatment, but also for understanding the biological functions and evolutionary adaptation of S. consticta and other benthic bivalves. Meanwhile, the obtained genome greatly improves our understanding of the genetics of molluscs and facilitates comparative evolutionary research.
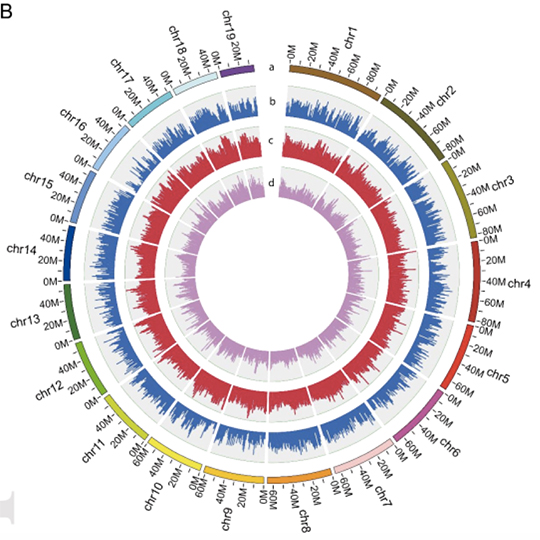
The Reference Genome Sequence of Scutellaria baicalensis Provides Insights into the Evolution of Wogonin Biosynthesis
Background:
Scutellaria baicalensis Georgi is important in Chinese Traditional Medicine where preparations of dried roots, ‘Huang Qin’, are used for liver and lung complaints including complementary cancer treatments. Recent studies have reported the pharmacological activities of preparations of Scutellaria roots including those of novel flavonoids. Understanding the genes responsible for biosynthesis of the various flavonoids made in S. baicalensis and their regulation, will lay a foundation for molecular breeding for improved, sustainable production.
Sampling & Sequencing Strategy:
Sample Preparation:
• Fresh leaf tissues from single-living black pepper plants
• Library preparation:
• Illumina short insert DNA library
• PacBio DNA SMRTbell library
• 10X Genomics DNA library
• Hi-C library
Sequencing: Illumina platforms and PacBio Sequel System
Results & Conclusion:
386.63 Mb of the 408.14 Mb genome were assembled, the sequences were sorted into 9 pseudochromosomes with a super-N50 of 33.9 Mb (Figure. 3). This study not only provides significant insight into the evolution of specific flavone biosynthetic pathways in members of the mint family, Lamiaceae, but also would facilitate the development of tools for enhancing bioactive productivity by metabolic engineering in microbes or by molecular breeding in plants.