概要
Using de novo sequencing to obtain the genomic information of bacterium or fungi (microbes) provides a fresh start for exploring the genetic structure and functions, studying the evolutionary origin of microbial populations, as well as developing potential applications of these abundant microbes in medicine, disease, agriculture, and environment. Novogene offers de novo sequencing service using both PacBio and Illumina platforms. We provide multifaceted sequencing services including genome survey, frame map, complete map, and fine map tailored to different research needs. For each project, our scientists will design the best sequencing strategy utilizing an optimal combination of short reads and long-range sequencing information to achieve the most comprehensive de novo assembly results for your genome of interest.
Service SpecificationsApplications
For individual research:
- Virulence research
- Drug resistance mechanism
- Molecular markers
For population research:
- Evolution relationship
- Population size
Advantages
- Highly experienced: We have completed numerical microbial sequencing projects with 20 publications on top tier journals.
- Largest sequencing capacity: We have the largest Illumina and PacBio sequencing capacities in the world, allowing us to provide high quality data, fast turnaround, and affordable prices.
Sample Requirements
| Sample Type | Amount (Qubit®) | Purity |
| Genomic DNA (For Illumina Platform) |
≥ 1 μg | OD260/280=1.8-2.0 |
| Genomic DNA/HMW DNA (For PacBio Platform) |
≥ 10 μg (Concentration ≥ 100 ng/μL) |
Sequencing Parameters And Analysis Contents
| Platform Type | Illumina Hiseq | PacBio Sequel | |
|
Read Length
|
Paired-end 150 bp
|
≥ 10K bp or 20K bp | |
| ≥ 100X for bacterial genomes | |||
| Recommended Sequencing Depth | ≥ 50x for bacterial and fungal genomes | ≥ 50X for fungal genomes | |
| Data Quality | Guaranteed ≥ 80% bases with Q30 or higher | Contig N50 ≥ 20K, Scaftig N50 ≥ 1M | |
|
Data Analysis
|
|
|
|
Note: For detailed information, please refer to the Service Specifications and contact us for customized requests.
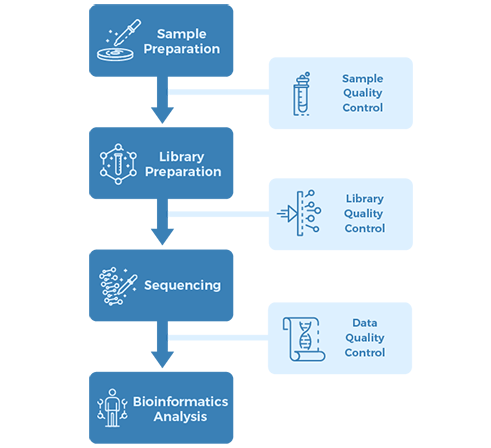
Project Workflow
Sampling:
DNA of R01-1 (ancestral strain) and B88-022 (closely related non-toxic strain)
Sequencing Strategy:
Illumina platform, paired-end 150 bp
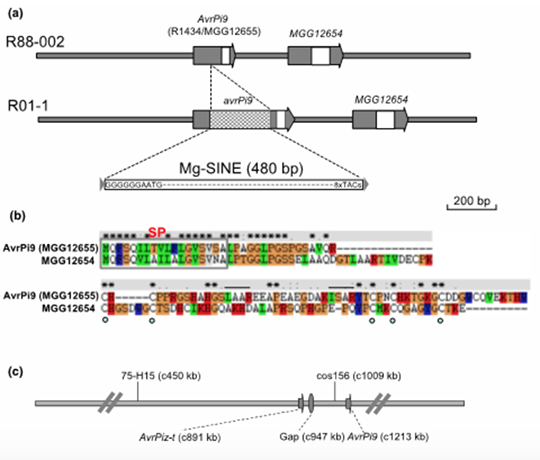
Conclusions:
Virulent isolate strains contain Mg-SINE within the AvrPi9 coding sequence. Loss of AvrPi9 did not lead to any discernible defects during growth or pathogenesis in M. oryzae. The prevalence of AvrPi9 correlates well with the avirulence pathotype in diverse blast isolates from the Philippines and China, thus supporting the broad-spectrum resistance conferred by Pi9 in different rice growing areas. Our results revealed that Pi9 and Piz-t at the Pi2/9 locus activate race specific resistance by recognizing sequence-unrelated AvrPi9 and AvrPiz-t genes, respectively.
Comparison of single-molecule sequencing and hybrid approaches for finishing the genome of Clostridium autoethanogenum and analysis of CRISPR systems in industrial relevant Clostridia
Background:
Clostridium autoethanogenum strain JA1-1 (DSM 10061) is an acetogen capable of fermenting CO, CO2 and H2 (e.g. from syngas or waste gases) into biofuel ethanol and commodity chemicals such as 2,3-butanediol. A draft genome sequence consisting of 100 contigs has been published.
Sampling:
DNA extracted from C. autoethanogenum strain JA1-1 was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) culture collection (DSM 10061).
Sequencing Strategy:
Illumina platform and PacBio platform
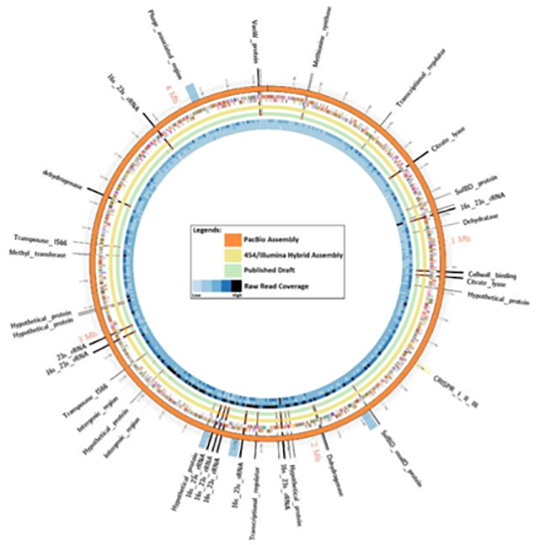
Results:
A comparative genomic analysis revealed short-read technologies were unable to overcome C. autoethanogenum DSM 10061 repeat regions largely associated with nine copies of the rRNA gene operons. The relatively low cost to generate the PacBio data (approximately US$1,500) and the outcome of this study support the assertion this technology will be valuable in future studies where a complete genome sequence is important and for complex genomes that contain large repeat elements.
Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi
Background:
Basidiomycota (basidiomycetes) make up 32% of the described fungi and include most wood-decaying species, as well as pathogens and mutualistic symbionts. Prior genomic comparisons suggested that the two decay modes can be distinguished based on the presence or absence of ligninolytic class II peroxidases (PODs), as well as the abundance of enzymes acting directly on crystalline cellulose (reduced in brown rot). Our results suggest a continuum rather than a dichotomy between the white-rot and brown-rot modes of wood decay.
Sampling:
DNA extracted from B. botryosum, G. marginata, and J. argillacea strains
Sequencing Strategy:
Illumina platform, paired-end 150 bp
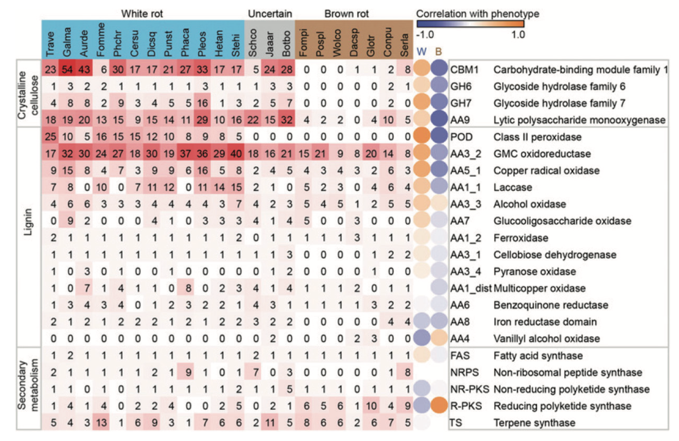
Conclusions:
Overall, the tree topology is consistent with prior studies using genome-scale datasets as well as phylogenetic analyses using small numbers of genes. Thirty-three basidiomycete genomes were searched for genes whose protein products are implicated in the breakdown of the polysaccharide portion of plant cell walls (cellulose, hemicellulose, pectin) using the CAZy database pipeline. The number of genes in each CAZy family, in each organism, is presented.
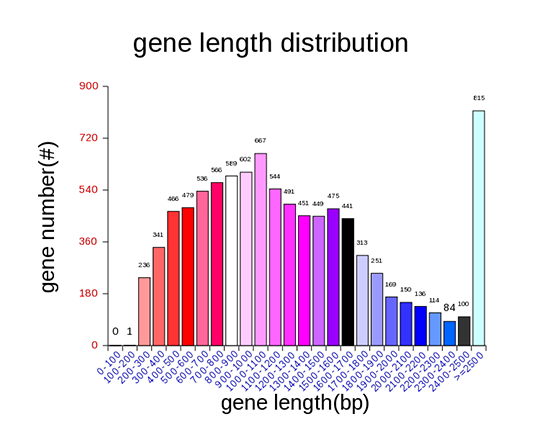
Gene length distribution
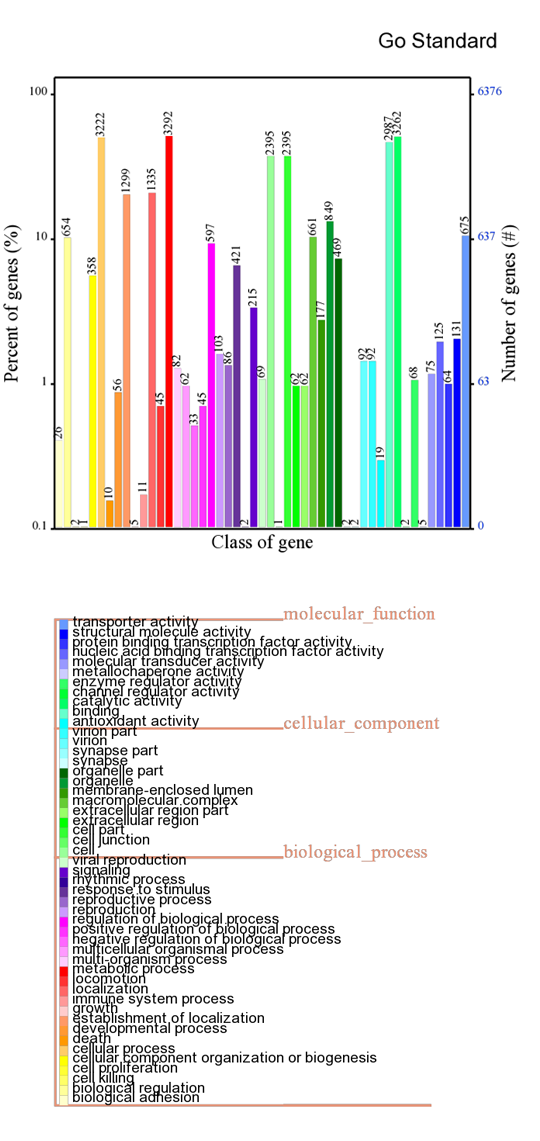
GO annotation
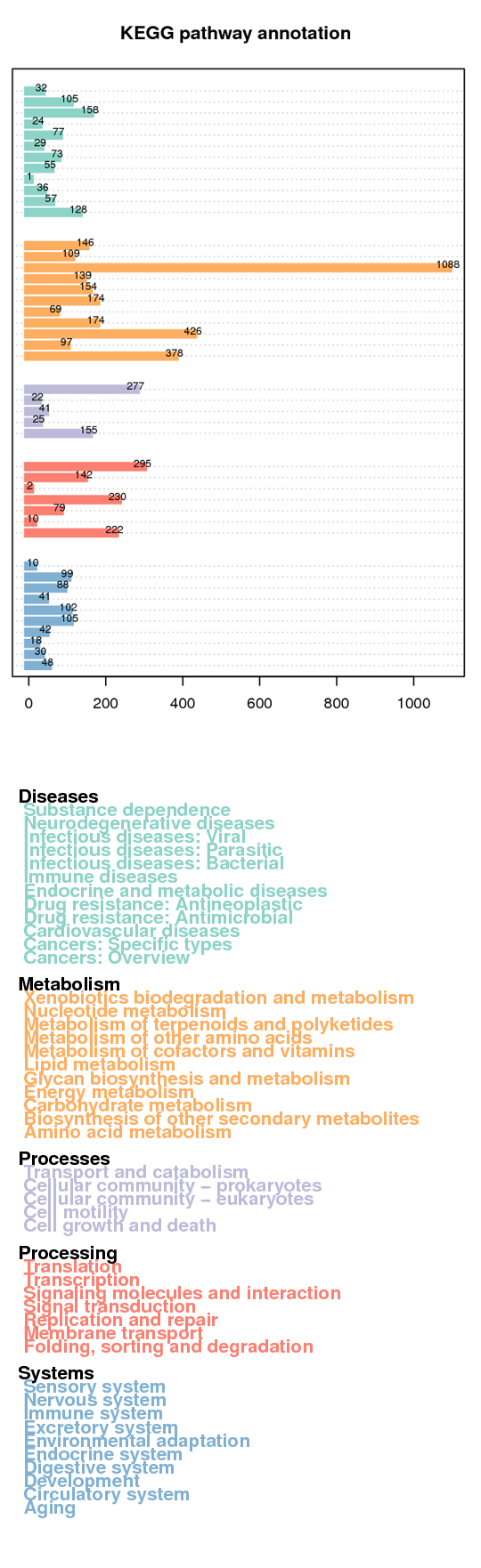
KEGG annotation
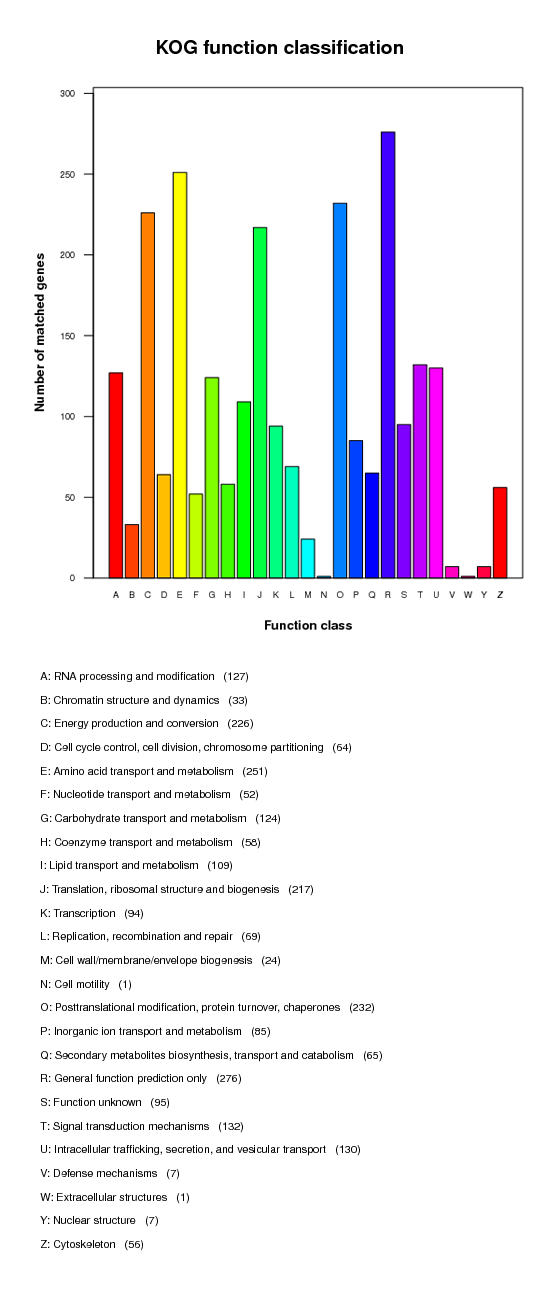
COG annotation
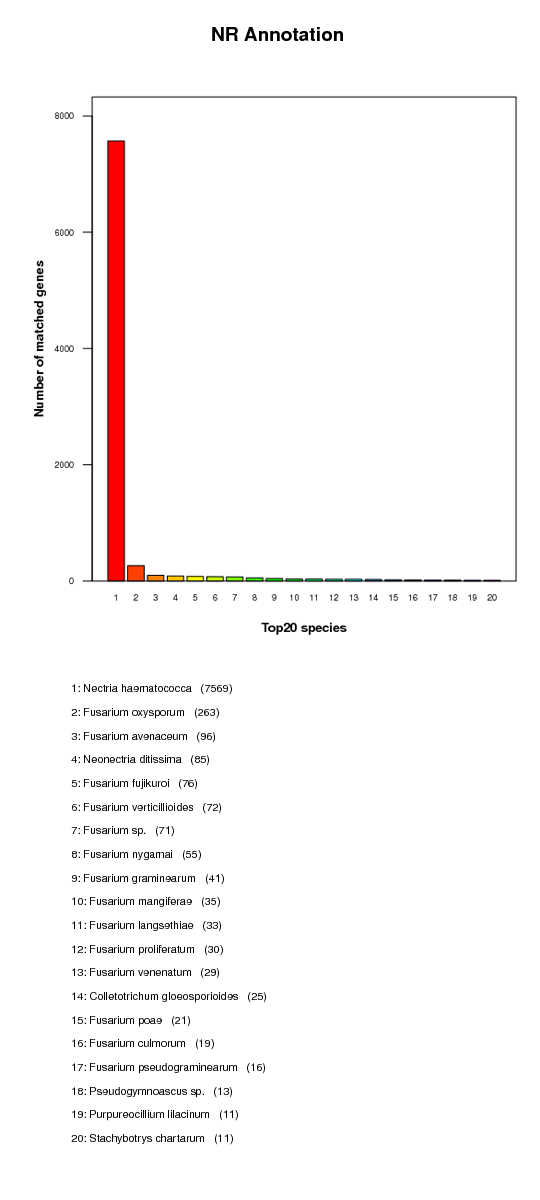
KR annotation
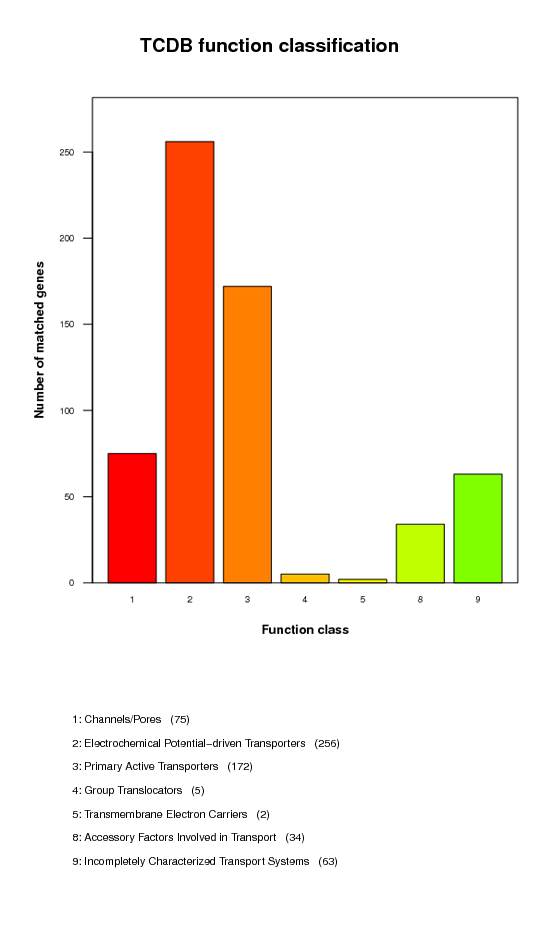
Transporter Classification Database (TCDB) function classification