動物・植物全ゲノムシーケンス(WGS) は、動物と植物それぞれの全ゲノムをシーケンスするために一般的に採用されている器械技術でありSNP, InDel, CNV, and SV. などのゲノム変異を同定することを目的としています。全ゲノムシーケンスは、一塩基レベルでの遺伝情報全体を決定する理想的なアプローチです。
ノボジーンは、超高速な納期、高品質のシーケンスデータ、信頼性の高い結果を備えた動物および植物の全ゲノムシーケンスサービスを提供しています。動物および植物の全ゲノムシーケンスは、集団遺伝学研究、ゲノムワイド関連研究(GWAS)、農業育種プログラムなど、さまざまな分野で応用されています。

動植物全ゲノムシーケンスの応用例
- 育種および種分化に関連する分子メカニズムを明らかにする
- 種の起源と進化
- 遺伝子開発を加速するためのリソースを提供
- 集団間で共通する遺伝的変異を特定
動植物全ゲノムシーケンスの利点
- 厳密なSOPに従った迅速なプロジェクト実施
- Q30≥85%の卓越したデータ品質
- 集団およびゲノムワイド関連研究にカスタマイズされた高度なソリューションが利用可能
- 植物や、ブタ、マウス、トラ、ミツバチなど幅広い動物での豊富な経験
動植物WGSの仕様:DNAサンプル要件
| プラットフォームタイプ | サンプルタイプ | サンプル量 (Qubit) | 純度 |
| Illumina
NovaSeq X Plus /NovaSeq6000 |
Genomic DNA | ≥ 200 ng | A260/280=1.8-2.0;
no degradation, |
| Genomic DNA
(PCR free non-350bp) |
≥ 5 μg | ||
| Genomic DNA
(PCR free -350bp) |
≥ 1.2 μg | ||
| PacBio Sequel II DNA CLR library |
HMW Genomic DNA | ≥ 8 μg | A260/280=1.8-2.0; A260/230=1.5-2.6; *NC/QC=0.95-3.00 Fragments should be ≥ 40 kb |
| PacBio Revio/sequel II/sequel IIe DNA HiFi library | HMW Genomic DNA | ≥ 5 μg | A260/280=1.8-2.0; A260/230=1.5-2.6; *NC/QC=0.95-3.00 Fragments should be ≥ 30 kb |
| Nanopore PromethION | HMW Genomic DNA | ≥ 8 μg | A260/280=1.8-2.0; A260/230=1.5-2.6; Fragments should be ≥ 30 kb |
*NC/QC:ナノドロップ濃度/キュービット濃度
動植物WGSの仕様:シーケンスおよび解析
| プラットフォームタイプ | Illumina NovaSeq X Plus /NovaSeq6000 | PacBio Revio/sequel II/sequel IIe | Nanopore PromethION |
| リード長 | Paired-end 150 bp | Average > 15 kb | Average > 17 kb |
| 推奨シーケンス深度 | For SNP/InDel detection: ≥ 10× | For SV detection: ≥ 20× | |
| For SV/CNV detection: ≥ 20× | |||
| データ解析 | Standard Analysis
Advanced Analysis
|
Standard Analysis
|
|
動植物ゲノムシーケンスサービスのワークフロー
サンプルおよびライブラリーの調製、ショートリードおよびロングリードシーケンス、データ品質管理からバイオインフォマティクス解析に至るまで、ノボジーン社は高品質の製品と専門的なサービスを提供します。各ステップは、高い科学水準と綿密な設計に基づき実施され、高品質の研究結果を保証します。




動植物全ゲノムシーケンスに関する論文
-
Resequencing of 1,143 indica rice accessions reveals important genetic variations and different heterosis patterns
Journal: Nature CommunicationsDate: 22 September, 2020IF: 12.121DOI:https://www.nature.com/articles/s41467-020-18608-0
-
Journal:Plant Biotechnology JournalIssue date: 18 September, 2020IF: 8.154DOI: https://onlinelibrary.wiley.com/doi/10.1111/pbi.13480
-
Journal: Molecular Plant date: 5 September, 2020IF:12.084DOI:
https://www.cell.com/molecular-plant/fulltext/S1674-2052(20)30296-3?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com
%2Fretrieve%2Fpii%2FS1674205220302963%3Fshowall%3Dtrue -
Journal:Nature GeneticsIssue date: 2018IF: 27.959DOI: https://www.nature.com/articles/s41588-018-0119-7
-
Genome re-sequencing reveals the evolutionary history of peach fruit edibility
Journal: Nature CommunicationsIssue date: 2018IF:12.353DOI:https://www.nature.com/articles/s41467-018-07744-3
デモデータ
シーケンス品質分布
シーケンス品質分布は、不正確な塩基が異常に多く組み込まれている領域を検出するために、すべての配列の全長にわたって調べられます。詳細なシーケンス品質分布は以下の通りです。
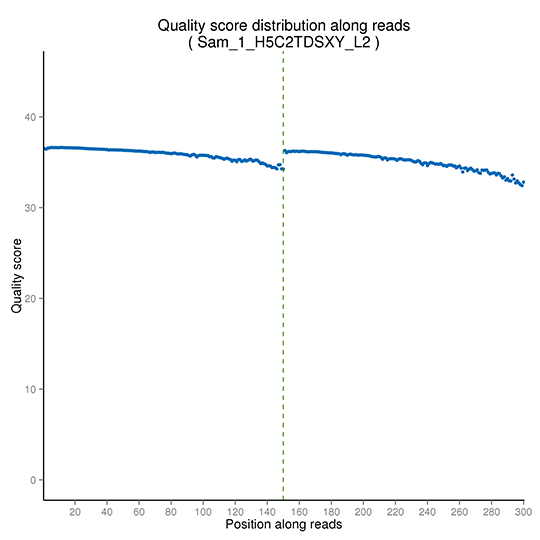
シーケンス品質分布
注:X軸はシーケンスリード内の塩基位置を示し、Y軸は各位置における全リードの平均phredスコアを示します。(ペアエンドシーケンスデータは一緒にプロットされ、最初のPE150 bpはリード1、次のPE150 bpはリード2を表します。)
シーケンスエラー率
シーケンスエラー率は、ディープシーケンスによる低頻度変異の正確な検出のため主要な交絡因子です。これはシーケンスデータの質を決定します。シーケンスエラー率は、シーケンスサイクルと大きく関連しており、イルミナの高スループットシーケンスプラットフォームの一般的な特徴である化学試薬の消費により、各リードの終わりに向かって上昇します。
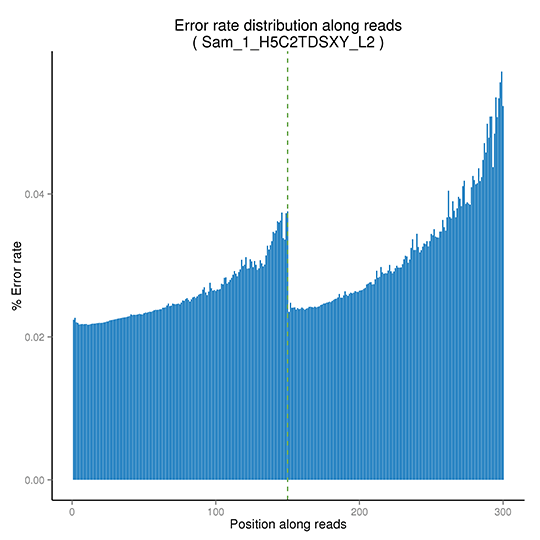
シーケンスエラー率分布
注:X軸は各シーケンスリードに沿った塩基の位置、Y軸はベースエラー率を示しています。(ペアエンドシーケンスデータは一緒にプロットされ、最初のPE150 bpはリード1、次のPE150 bpはリード2を表します。 )
アダプターを含む、または低品質のリードのフィルタリング
シーケンス後のリードには、低品質のリードやアダプターを含むリードが含まれることが多く、これは下流の解析の品質に影響します。これを避けるためには、リードをフィルターしてクリーンなリードを得る必要があります。リードのフィルタリングは以下の条件で行います:
(1) ペアリードのどちらかにアダプターの混入がある場合、ペアリードを除去する;
(2) 不確かなヌクレオチド(N)がどちらかのリードの10%以上を占める場合、ペアリードを除去する;
(3) 低品質ヌクレオチド(ベースの品質が5未満、Q≦5)がどちらかのリードの50%以上を占める場合、ペアリードを除去する。
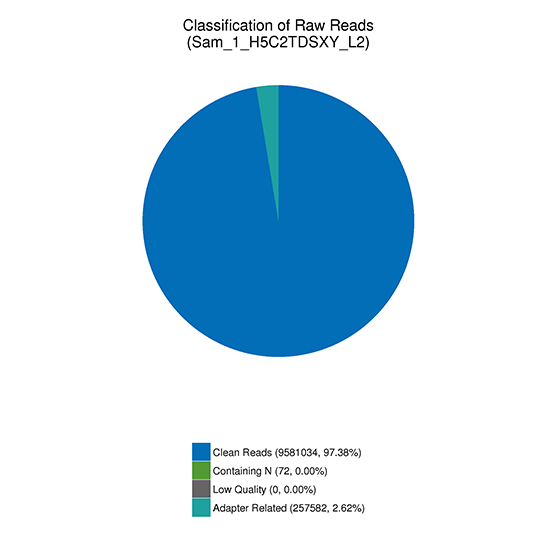
注:シーケンスしたリードの分類
(1) アダプター関連:(アダプターを含むリード)/(全リード)
(2) Nを含む:(10%以上のNを含むリード)/(全リード)
(3) 低クオリティ: (低品質のリード) / (全リード)
(4) (4)クリーンリード: (クリーンリード) / (全リード)
参照ゲノムとのアライメント
参照ゲノムとのマッピング統計
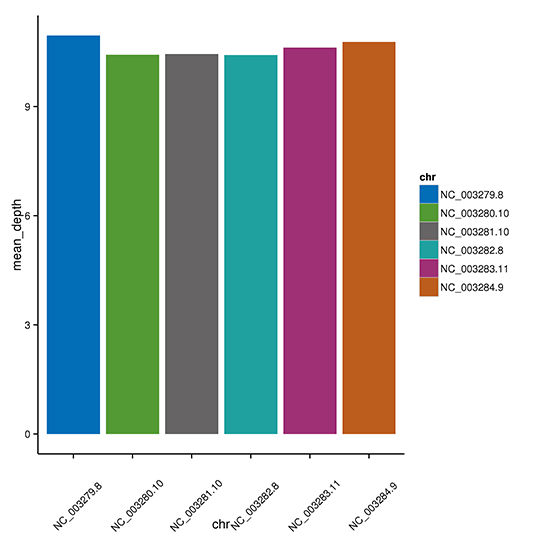
各染色体の平均デプス
注:X軸は染色体、Y軸は平均デプスを示しています。
標準解析
SNP検出とアノテーション
ANNOVARは、ゲノムから発見された遺伝子変異を、現在の情報を利用して効率的にアノテーションするソフトウェアです。参照ヌクレオチド、開始位置、終了位置、観測ヌクレオチド、染色体を含むバリアントのインデックスが提供され、ANNOVARで遺伝子ベースのアノテーション、領域ベースのアノテーション、フィルターベースのアノテーション、その他の機能を実行することができます。
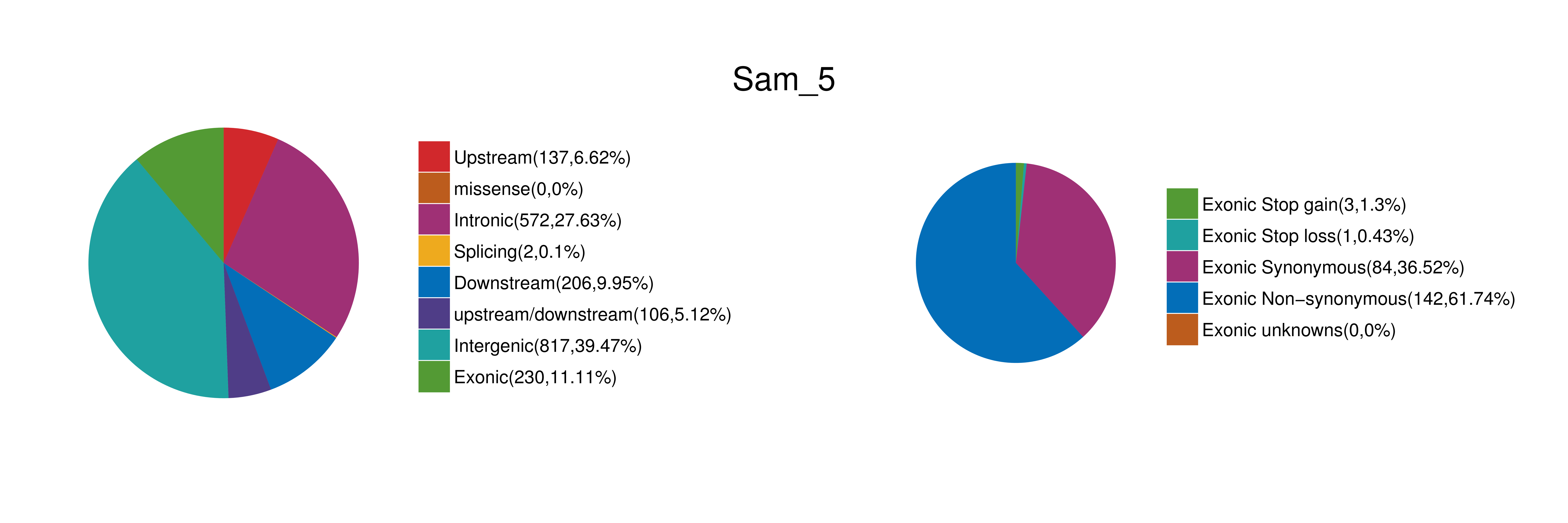
注:図は(A)ゲノムの異なる領域におけるSNP数(左)と(B)コーディング領域における異なるタイプのSNP数(右)の分布を示しています。
SNPの品質分布
検出されたSNPの信頼性を評価するために、サポートリード数、SNP品質、隣接SNP間の距離の分布を確認しました。結果を以下に示します。
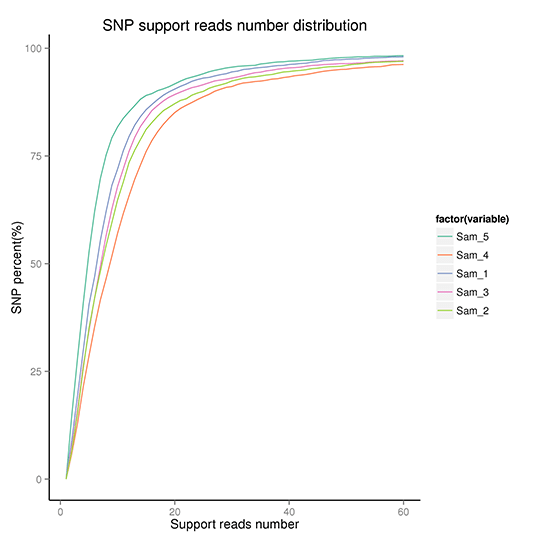
SNPクオリティの累積分布
注:これらの図は、SNPの品質分布、SNPサポートリード数の分布、隣接SNP間の距離分布、SNPクオリティの累積分布を示す。
SNP変異頻度
T:A>C:G変異を例にとると、このカテゴリーにはTからCへの変異とAからGへの変異が含まれます。T>C変異が二本鎖のどちらかに現れると、もう一方の鎖の同じ位置にA>G変異が見つかります。したがって、T>C変異とA>G変異は1つのカテゴリーに分類される。これにより、全ゲノムSNP変異は6つのカテゴリーに分類されます。
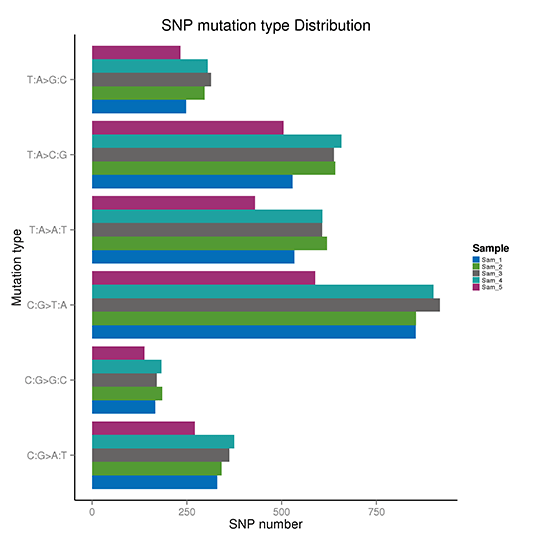
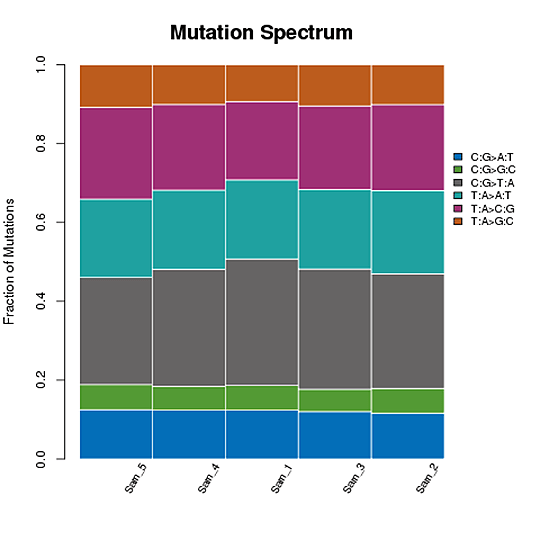
SNP変異の頻度
注:X軸はSNPの数、Y軸は変異のタイプを示しています。
CNVの検出とアノテーション
コピー数変異(CNV)は、DNA断片が参照ゲノムと比較して様々なコピー数で存在する場合に起こる構造変異の一種です。ゲノム中の欠失や重複をピンポイントで検出します。リファレンスゲノムのリードデプスに基づき、CNVnator は以下のパラメータ ‘-call 100’ で欠失や重複の可能性のある CNV を検出することができます。検出されたCNVは、ANNOVARによってさらにアノテーションされます。
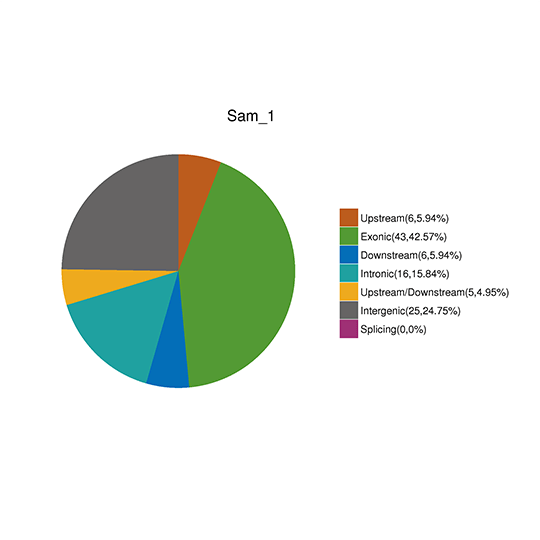
CNVアノテーション
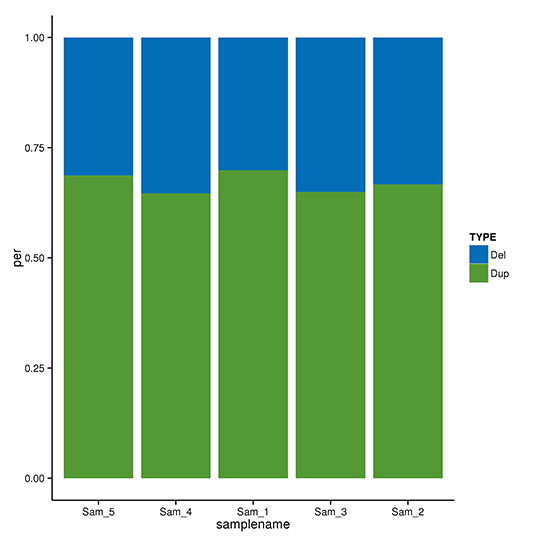
CNVのANN変異タイプ分布
注:X軸はサンプルを示し、Y軸は各タイプのCNVの割合を示しています。
*デモレポートをご希望の方は お問い合わせください