概要

Chromatin Immunoprecipitation Sequencing (ChIP-Seq) provides genome-wide profiling of DNA targets for histone modification, transcription factors, and other DNA-associated proteins. It combines the selectivity of chromatin immuno-precipitation (ChIP) to recover specific protein-DNA complexes, with the power of next-generation sequencing (NGS) for high-throughput sequencing of the recovered DNA. Additionally, because the protein-DNA complexes are recovered from living cells, binding sites can be compared in different cell types and tissues, or under different conditions. At Novogene, we provide high-quality sequencing and comprehensive bioinformatics solutions for your ChIP-Seq projects.
Service SpecificationsApplications
- Applications range from transcriptional regulation to developmental pathways to disease mechanisms and beyond.
Advantages
- Cost-effective: Rapid and efficient genome-wide profiling of multiple samples, using only 1/100 of the amount of DNA required for ChIP-chip.
- Unsurpassed data quality: We guarantee that ≥ 80% of bases have a sequencing quality score ≥ Q30, exceeding Illumina’s official guarantee of ≥ 75%.
- Comprehensive analysis: Expert bioinformatics analyses utilizing widely accepted MACS2 software and latest programs for motif prediction, peak annotation, functional analysisand data visualization.
- Professional bioinformatics:A bioinformatics analysis team composed of Ph.D. scientists entirely for Chip-Seq data analysis.
Sample Requirements
| Sample Type | Required Amount | Fragment size | Purity |
|
Enriched DNA Sample
|
≥50 ng (Concentration ≥2ng/μL)
|
100 bp~500bp
|
OD260/280=1.8-2.0
|
Sequencing Parameter and Analysis
| Platform | Illumina Novaseq 6000 |
| Read length | Pair-end 150 |
| Recommended Sequencing Depth | ≥ 20 million read pairs per sample for the species with reference genome |
| Standard Data Analysis |
|
Note: For detailed information, please refer to the Service Specifications and contact us for customized requests.
Project Workflow
Sampling & Sequencing Strategy:
2.1. Samples:
CaSki-pHAGE cells, CaSki-KDM5C cells
2.2. Library preparation:
ChIP-seq library and RNA-seq library
2.3. Sequencing:
Illumina HiSeq
2.4. Bioinformatics analysis:
ChIP-seq standard analysis and gene expression analysis
Results:
1) Whole genome ChIP-seq revealed existence of the EGFR and c-MET super-enhancers in the human cervical cancer cell line
The existence of possible super-enhancers was screened by ChIP-seq analysis of H3K27Ac in the CaSki cells, as well as the CaSki-vector control and CaSki-KDM5C cells. It was found that there the two protooncogenes, EGFR and c-MET, each contained a super-enhancer. The EGFR super-enhancer is located in the first intron while the c-MET super-enhancer resides within the c-MET gene 50 region through intron 2 (Figure 1).
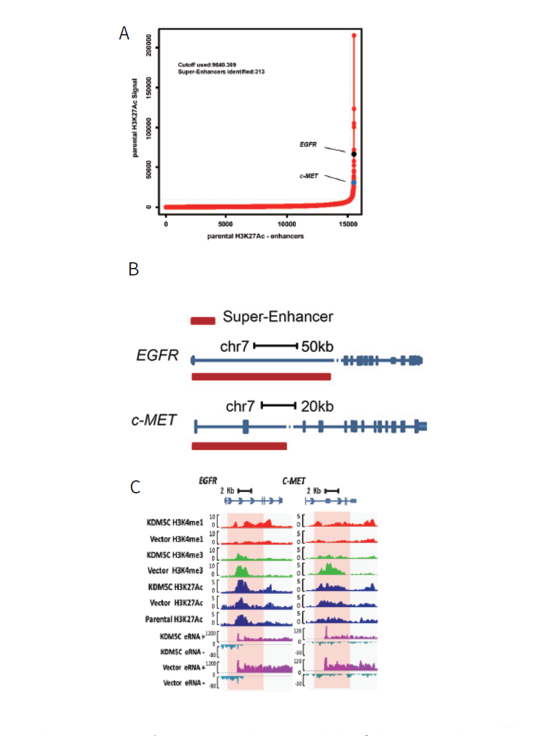
2) KDM5C regulates cervical cancer cell EGFR and c-MET expression by modulating their super-enhancer H3K4 methylation dynamics
ChIP-qPCR of KDM5C was performed and displayed a direct correlation between the restoration of KDM5C and its enrichment. KDM5C restoration led to specific increased H3K4me1 in global super-enhancers rather than in adjacent regions, confirming KDM5C as a specific enhancer regulator (Figure 2).
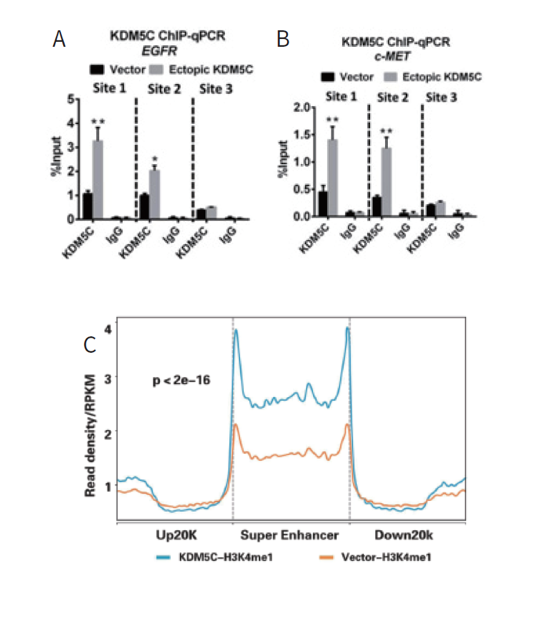
Figure 2. KDM5C were enriched at super-enhancer or other regions of EGFR and c-MET.
Conclusion:
This study generated a carcinogenic model of HPV infection: HPV16 E6 binds to KDM5C and form an E6‒E6AP‒KDM5C complex, thereby degrading KDM5C in a polyubiquitin-dependent manner. As a result, the super-enhancers of key protooncogenes, EGFR and c-MET, become highly upregulated, increasing their expressions and promoting tumor cell growth. This finding has provided novel insights into virus-induced cancer.
Reference: Chen X, Loo J X, Shi X, et al. E6 Protein Expressed by High-Risk HPV Activates Super-Enhancers of the EGFR and c-MET Oncogenes by Destabilizing the Histone Demethylase KDM5C[J]. Cancer Research, 2018, 78(6): 1418-1430.
Figure 1 Distribution of MAPQ
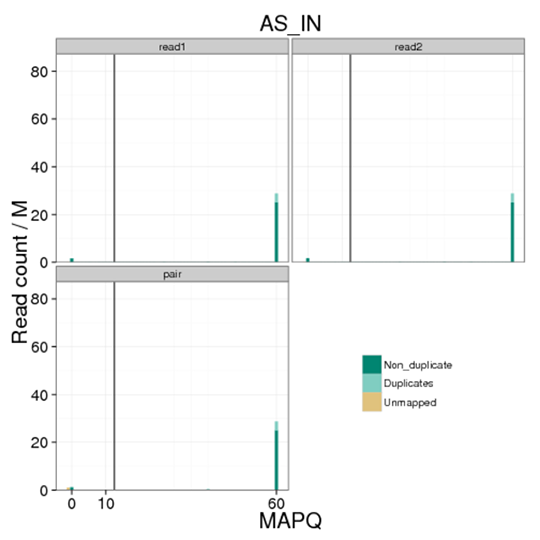
Figure 2 Plots of strand cross correlation
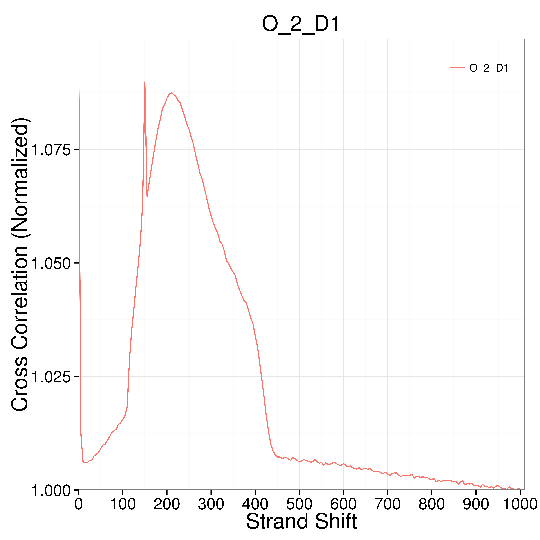
Figure 3 Distance distribution of peaks to TSS
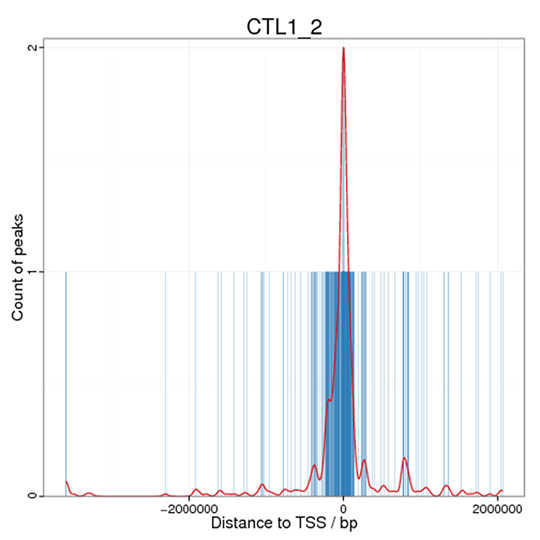
Figure 4 Motif analysis
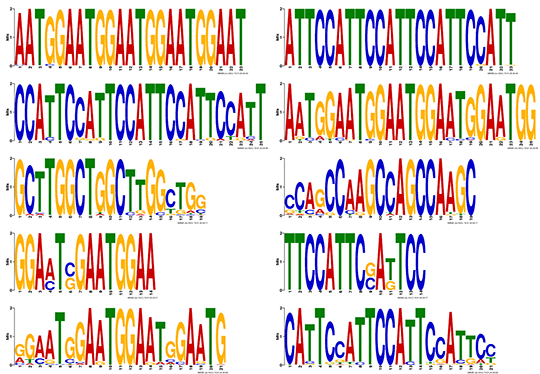
Figure 5 Peak distribution in functional gene region
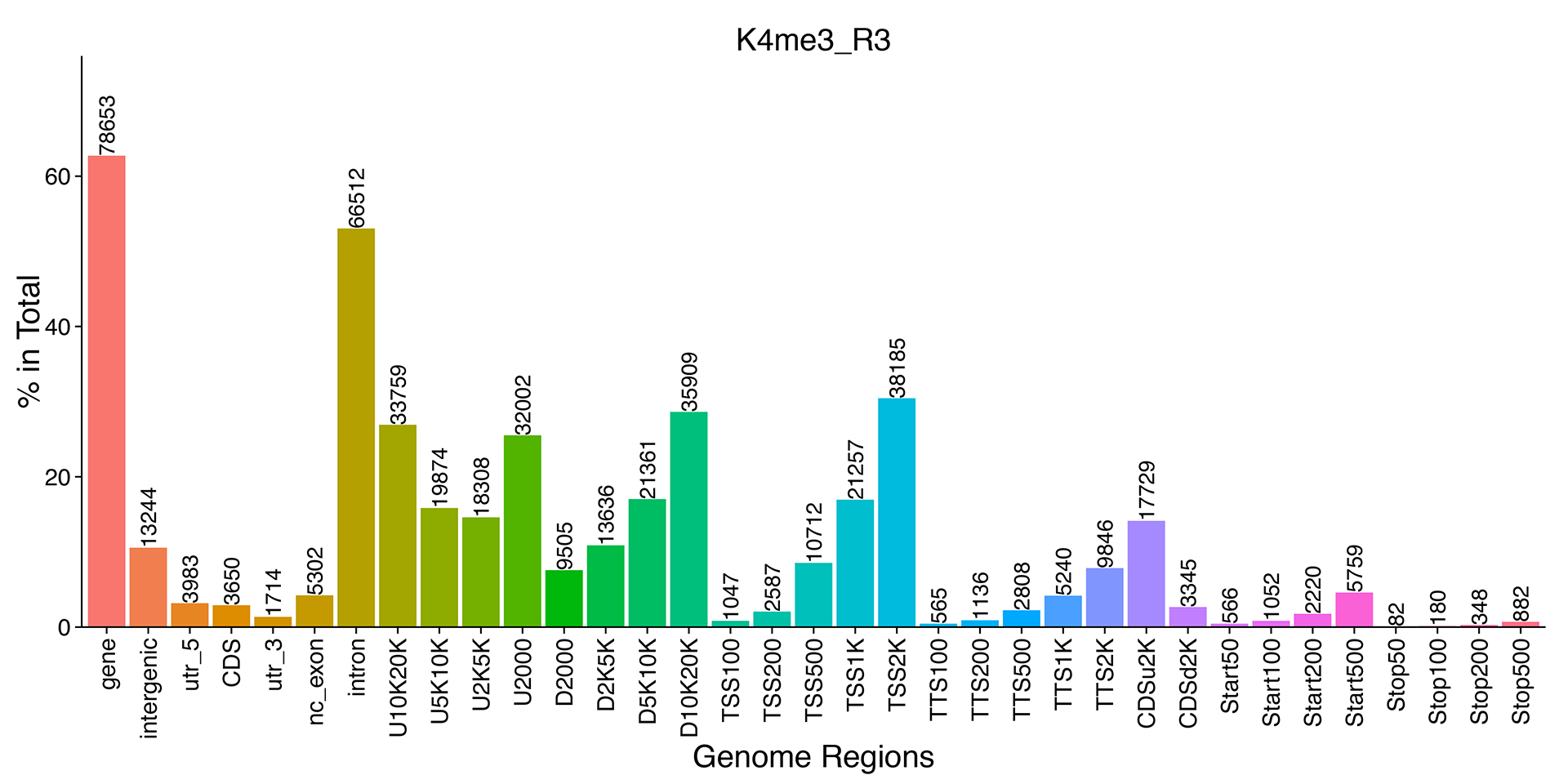
Figure 6 GO enrichment
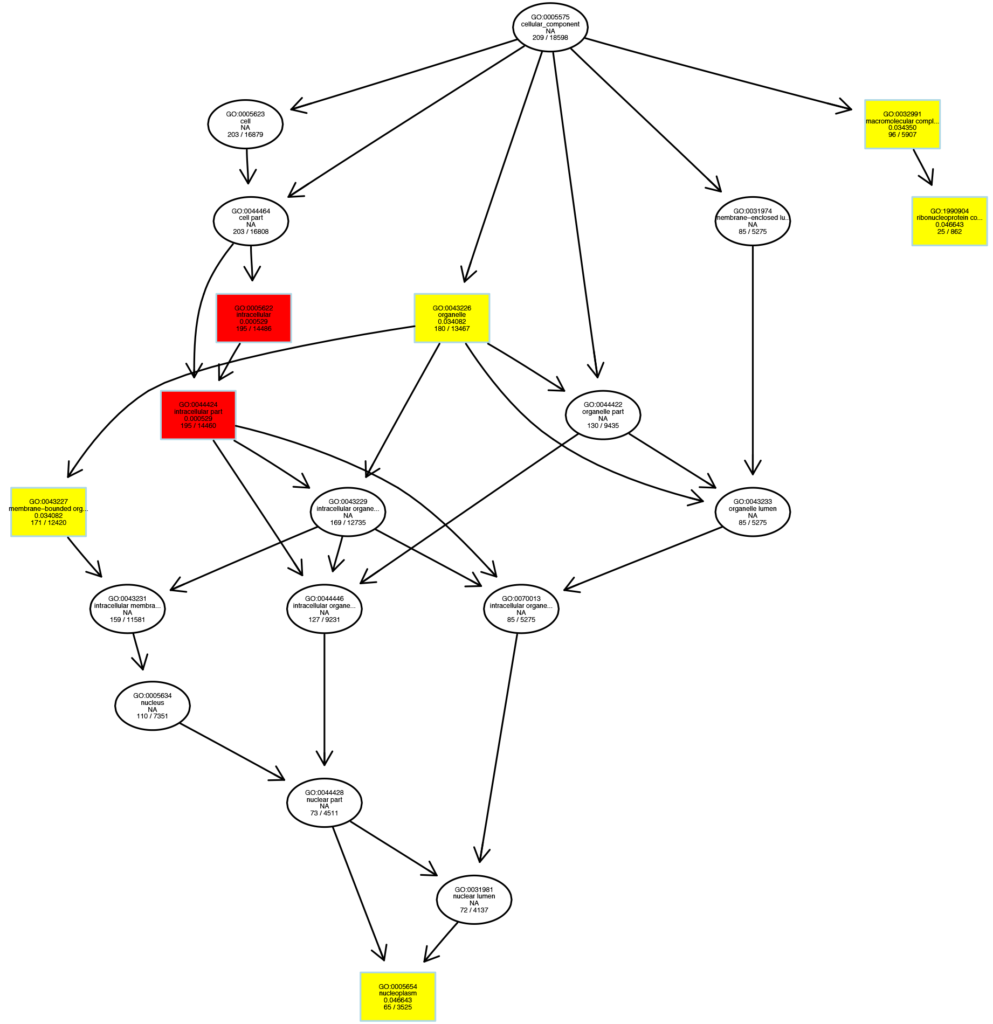
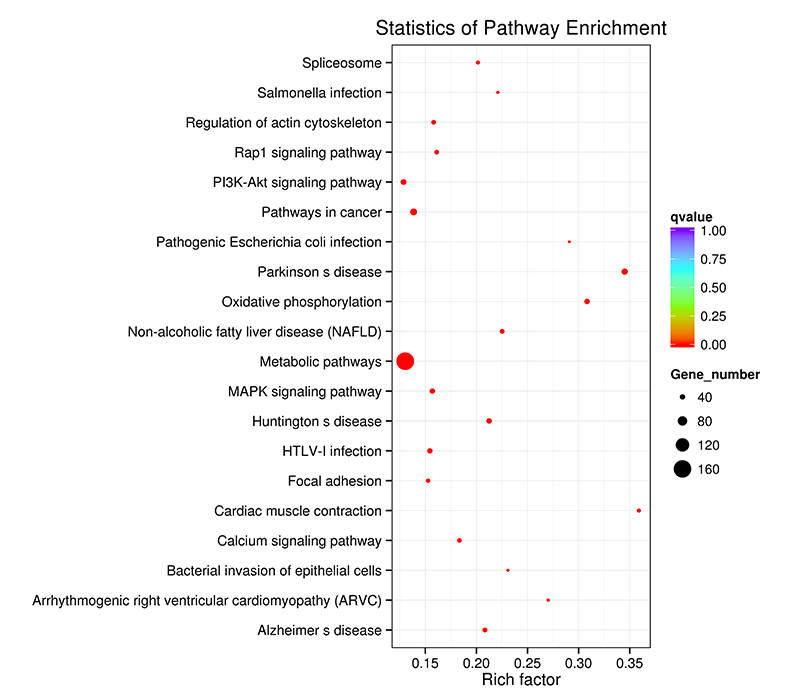
Figure 7 KEGG enrichment scatter