微生物全ゲノムシーケンス の紹介
微生物全ゲノムシーケンスは、微生物ゲノム全体の塩基配列を決定するだけでなく、複数の参照ゲノムと新しい生物のマッピングされたゲノムを比較するためにも重要なアプローチです。細菌、ウイルス、その他の微生物ゲノムをシーケンスすることは、正確な参照ゲノムの作成、微生物の同定、その他の比較ゲノム研究にとって重要です。
PCRのような従来のアプローチと比較して、全ゲノムシークエンシングは労力を要するクローニングやマッピングのステップを必要としません。したがって、時間とコスト効率に優れています。さらに、このハイスループットシーケンスアプローチは、マルチプレックスの恩恵により、多数のサンプルのシーケンスを同時に行うことができます。
微生物全ゲノムシーケンスのアプリケーション
- ターゲットゲノム内の変異の検出が可能
- 個体差の解釈
- 大規模な進化研究が可能
- 新規種同定の前提研究が可能
微生物全ゲノムシーケンスの利点
- 豊富な経験:病原性細菌、プロバイオティクス、食用細菌、医薬用菌株、工業用菌株など、幅広い分野をカバーする著名なプロジェクトを成功させてきました。
- プロフェッショナルなサービス:材料の選択、ライブラリ構築、シークエンスからデータ解析まで、各ステップにおいて科学的かつ綿密な設計を行い、高品質な研究結果をお約束します。
- 包括的な解析:株参照ゲノムのSNP、InDel、SVなどの変異情報を検出し、種の進化、集団特性、選択圧などの研究を進める。変異と差異をワンストップで解析します。
- 厳密な品質管理:サンプル検証を行うことで、シーケンスデータの品質を保証します。
- 高品質ライブラリ調製:データの質を保証するため、すべてのライブラリーはインサートサイズを最適化するようサイズ選択されています。
微生物全ゲノムシーケンス仕様:DNAサンプル要件
| ライブラリータイプ | サンプルタイプ | サンプル量 (Qubit) | 液量 | 濃度 | 純度(NanoDrop™/Agarose Gel) |
| Microbial whole genome library (350bp) | Genomic DNA | ≥ 200 ng | ≥ 20 μL | ≥10 ng/μL | A260/280=1.8-2.0; 分解なし, コンタミなし |
| Microbial whole genome library(PCR-free 350bp) | Genomic DNA | ≥ 1.2 μg | ≥ 20 μL | ≥10 ng/μL |
微生物全ゲノムシーケンス仕様:シーケンスパラメーターおよび解析項目
| シーケンスプラットフォーム | Illumina NovaSeq 6000 |
| リード長 | Paired-end 150 bp |
| 推奨シーケンス深度 | ≥ 100x for bacterial genomes |
| ≥ 50x for fungal genomes | |
| 標準データ解析内容 |
|
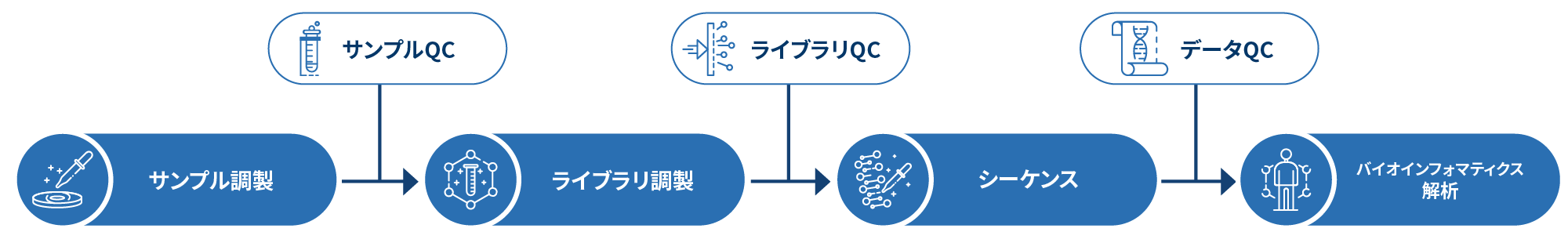
微生物全ゲノムシーケンスのワークフロー
プロジェクトワークフローの最初のステップでは、サンプル品質管理(サンプルQC)を行い、お客様のサンプルが微生物WGSの基準を満たしていることを確認します。次に、ターゲットとする生物に応じて適切なライブラリーを調製し、その品質を検査します(ライブラリQC)。次に、ペアエンド150bpシーケンス戦略を用いてサンプルのシーケンスを行い、得られたデータの品質を保証するためにデータ品質管理(Data QC)を行います。最後に、バイオインフォマティクス解析が行われ、論文発表可能な結果が提供されます。以下のフローシートでは、私たちの微生物WGSが従うステップバイステップのプロトコルを説明しています。
サンプルの調製に続いて、DNAライブラリの調製が行われ、品質と収量が確認されます。ゲノムDNAは350bpのサイズで断片化され、サンプル精製ビーズによってサイズが選択されます。選択された断片は末端を加工、A-tailが付与され、全長アダプターとライゲーションされます。サンプルのシーケンスにはイルミナPE150テクノロジーが採用され、最終段階ではバイオインフォマティクス解析が行われます。




微生物全ゲノムシーケンスに関する論文
-
Dynamics and Microevolution of Vibrio parahaemolyticus Populations in Shellfish Farms
mSystems Date: 12 January 2021IF: 6.663DOI: https://doi.org/10.1128/mSystems.01161-20
-
Science of the Total Environment Date: 20 june 2021IF:6.551DOI: https://10.1016/j.scitotenv.2021.145767
-
food chemistry Date: 15 November 2020IF: 6.306DOI: https://10.1016/j.foodchem.2020.127316
-
Whole genome sequence of Diaporthe capsici, a new pathogen of walnut blight
Genomics Date: 23 February 2021IF: 6.205DOI: https://doi.org/10.1016/j.ygeno.2020.04.018
-
Effect of steel slag in recycling waste activated sludge to produce anaerobic granular sludge
Chemosphere Date: 25 October 2020IF: 5.108DOI: https://doi.org/10.1016/j.chemosphere.2020.127291
-
Journal of Global Antimicrobial Resistance Date: 23 December 2020IF: 4.035DOI: https://10.1016/j.jgar.2020.08.002
-
Alterations of gut microbiota contribute to the progression of unruptured intracranial aneurysms
Nature Communications Date: 25 june 2020IF:14.919DOI: https://10.1038/s41467-020-16990-3
SNP変異頻度
一塩基多型(SNP)とは、一塩基の変異のことであり、一塩基の転移や転座を含め、ゲノム上の特定の位置で起こりえます。
例えば、T: T>C変異が二本鎖のどちらかに現れると、もう一方の鎖の同じ位置にA>G変異が見つかります。したがって、T>C変異とA>G変異は1つのカテゴリーに分類されます。それにより、全ゲノムSNP変異は6つのカテゴリーに分類できます。各タイプの頻度を図Xに示します。
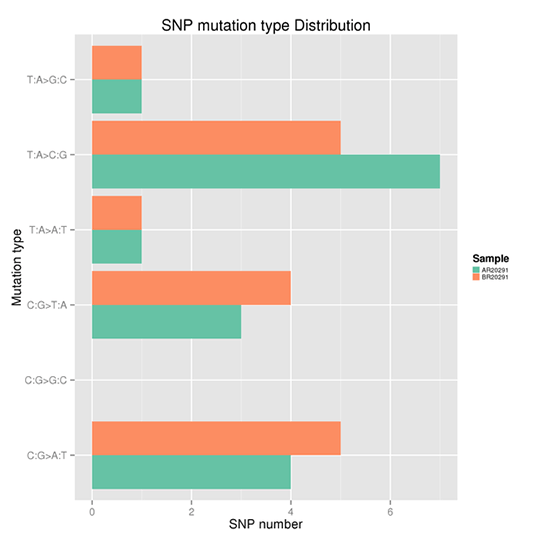
注:X軸はSNPの数、Y軸は変異のタイプを示します。
CDSに位置するInDelの長さ分布
InDelとはDNA中の50bp以下の配列の挿入または欠失を指します。結果は特定のInDelの長さにいくつかのピークが存在することを示しています。非フレームシフトInDelはフレームシフトInDelに比べてゲノムへの影響は小さくなります。
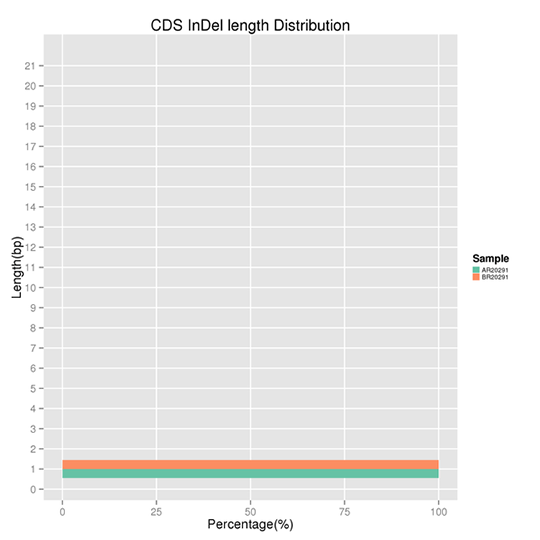
注:X軸はある長さのInDelの割合を表し、Y軸はInDelの長さを表します。
SVの長さ分布
構造変異(SV)とは、欠失、重複、挿入、逆位、平衡転座などの比較的大きなサイズ(>50 bp)の変異を持つゲノムの変異である。
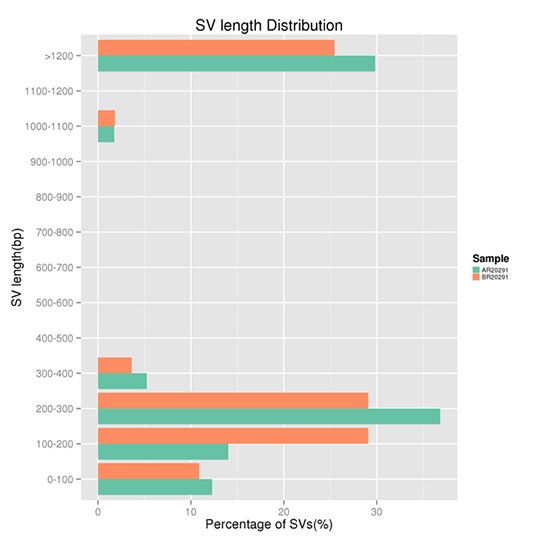
注:X軸はある長さの範囲のSVの割合、Y軸はSVのある長さの範囲を示します。なお、SVsの検出には、ライブラリ構築時のDNAインサートの長さが大きく影響します。
ゲノム上のCNVの分布
コピー数変異(CNV)は、DNA断片が参照ゲノムと比較して様々なコピー数で存在する場合に起こる構造変異の一種です。ゲノム上の欠失や重複をピンポイントで検出することができます。
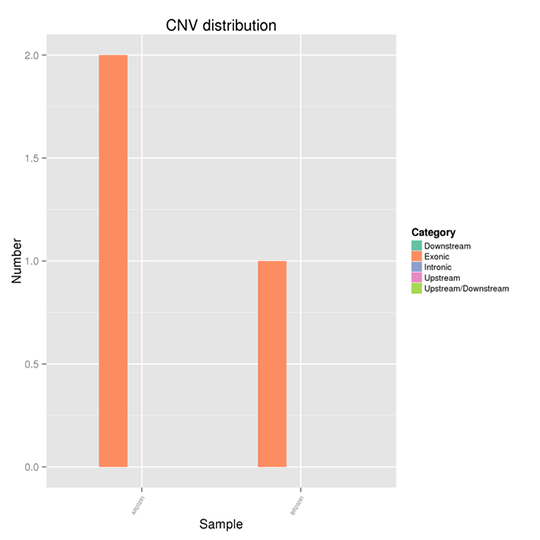
注:X軸はサンプルを表し、Y軸は異なる領域におけるCNVの数を示します。
変異の可視化
全ゲノムにおける構造的変異を適切に可視化するために、変異のタイプをCircosで示します:
(1)SNP/InDelタイプは、密度分布が描画されます。
(2)SV/CNVタイプは、位置とサイズが描画されます。
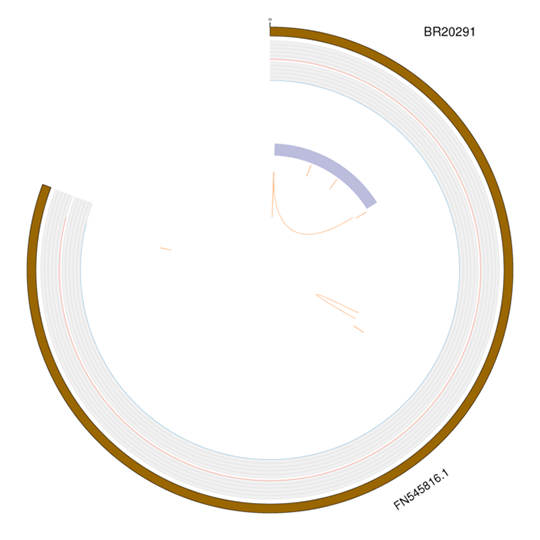
注:外側から内側へ:染色体、SNP、InDel、CNV重複、CNV欠失、SV挿入、SV欠失、SVインバージョン、SV ITX、SV CTX
*デモレポートをご希望の方はお問い合わせください。