概要

全エクソームシーケンス(WES)は次世代シーケンス技術(NGS)を採用しており、全ゲノムシーケンス(WGS)に代わるコスト効率の高い手法です。約180,000のエクソン(ゲノムのタンパク質コード領域)で構成されるヒト全エクソームは、ヒトゲノムのわずか1~2%を占めるに過ぎませんが、メンデル性疾患に関連する疾患関連変異の最大85%はこれらの領域で発生しています[1]。これらの領域をターゲットとすることで、ヒト全エクソームシーケンス(hWES) は、ゲノム変異、生殖細胞系列変異、体細胞変異、および発症メカニズムを示す詳細なシーケンスおよび解析アプローチを提供します。hWES サービスは、遺伝病関連変異、複雑な疾患、がん研究、またはヒト集団遺伝学など、研究者の幅広い研究をサポートします。
ノボジーンのhWESサービスは、高品質なデータ、研究目標の達成に役立つ論文発表可能な結果など、多くの利益をもたらす便利なソリューションを提供します。ノボジーンは、CLIA/CAP/ISO17025基準に準拠し、バリデートされた臨床グレードのシーケンスラボを備えており、臨床全エクソームシーケンスサービスで正確な診断を提供します。
ヒト全エクソームシーケンスの応用例
ヒト全エクソームシークエンスは、研究者がさまざまな最先端の研究および臨床上の疑問に対する答えを得るために役立っています:
- エクソン検出によるゲノム変異研究
- 研究および臨床におけるサンプルの病因メカニズムおよび分子特性解析
- ツールとしてのがん生検
ノボジーンのヒト全エクソームシーケンスの利点
- hWESはエクソーム領域に重点を置いているため、WGSと比較して、質の高いデータを大幅にデータ量を減らしながら、高いシーケンスデプスを達成することができます。 プロフェッショナルなサービス:材料の選択、ライブラリ構築、シークエンスからデータ解析まで、各ステップにおいて科学的かつ綿密な設計を行い、高品質な研究結果をお約束します。
- hWESは解析の感度を高め、希少変異の検出を容易にします。 厳密な品質管理:サンプル検証を行うことで、シーケンスデータの品質を保証します。
- ノボジーンの卓越した専門的バイオインフォマティクスパイプラインと国際的に認められたソフトウェアにより、お客様は常に信頼性が高く、論文発表に適したデータを得ることができます。
hWES仕様:DNAサンプル要件
| プラットフォームタイプ | サンプルタイプ | サンプル量 (Qubit®) | 純度 |
| Illumina NovaSeq 6000 | Genomic DNA | ≥ 300 ng | A260/280 = 1.8-2.0; No degradation, no contamination |
| cfDNA/ctDNA | ≥ 30 ng | ragments should be in multiples of 170 bp,with no genomiccontamination | |
| Genomic DNA from FFPE tissue | ≥ 500 ng | agments should be ≥ 1000 bp |
注:サンプル量の数値は参考までに記載しています。詳しくは、サービス仕様とサンプル要件をダウンロードしてください。詳細情報については、カスタマイズされたご要望をご連絡ください。
hWES仕様:シーケンスと解析
| シーケンスプラットフォーム | Illumina NovaSeq 6000 |
| リード長 | Paired-end 150 bp |
| 推奨シーケンス深度 | For Mendelian disorder/rare disease: effective sequencing depth above 50× (6G) |
| For tumor sample: effective sequencing depth above 100× (12G) | |
| 標準データ解析内容 |
|
注:表示されているシーケンスデプスおよび解析内容は参考です。詳しくはサービス仕様をダウンロードしてください。詳細な情報については、お客様のご要望をお聞かせください。
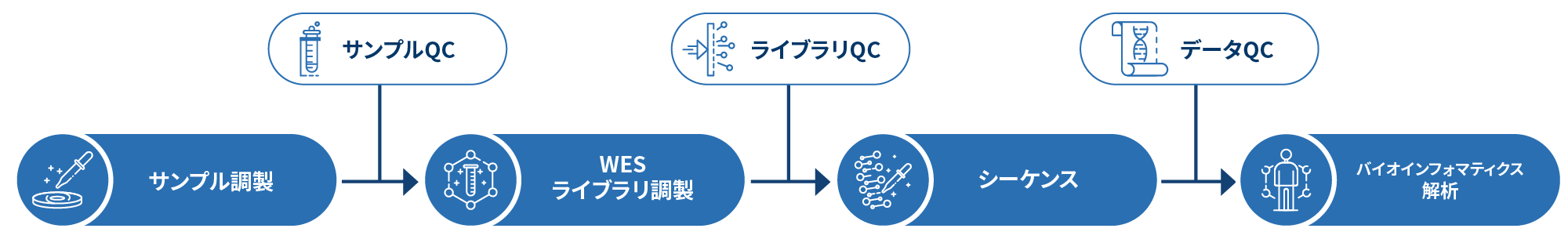
hWESサービスのプロジェクトワークフロー
サンプル調製、ライブラリー調製、DNAシーケンス、データQCからバイオインフォマティクス解析まで、ノボジーンは高品質なデータと専門的なサービスを提供します。各ステップは、高い科学水準と綿密な設計に基づき実施され、高品質の研究結果を保証します。




ヒト全エクソームシーケンスに関する論文
ヒト全エクソームシーケンス(hWES)により、研究者はまれなメンデル性疾患、複雑な疾患、がん、またはヒト集団研究におけるタンパクをコードするバリアントをコスト効率よく明らかにすることができます。ここでは、ノボジーンのhWESサービスを利用した優れた学術論文を要約しています。
-
Dynamics and Microevolution of Vibrio parahaemolyticus Populations in Shellfish Farms
mSystems Date: 12 January 2021IF: 6.663DOI: https://doi.org/10.1128/mSystems.01161-20
-
Science of the Total Environment Date: 20 june 2021IF:6.551DOI: https://10.1016/j.scitotenv.2021.145767
-
food chemistry Date: 15 November 2020IF: 6.306DOI: https://10.1016/j.foodchem.2020.127316
-
Whole genome sequence of Diaporthe capsici, a new pathogen of walnut blight
Genomics Date: 23 February 2021IF: 6.205DOI: https://doi.org/10.1016/j.ygeno.2020.04.018
-
Effect of steel slag in recycling waste activated sludge to produce anaerobic granular sludge
Chemosphere Date: 25 October 2020IF: 5.108DOI: https://doi.org/10.1016/j.chemosphere.2020.127291
-
Journal of Global Antimicrobial Resistance Date: 23 December 2020IF: 4.035DOI: https://10.1016/j.jgar.2020.08.002
-
Alterations of gut microbiota contribute to the progression of unruptured intracranial aneurysms
Nature Communications Date: 25 june 2020IF:14.919DOI: https://10.1038/s41467-020-16990-3
データQCシーケンスエラー率分布
シーケンスエラー率は、ディープシーケンスによる低頻度変異の正確な検出の主要な交絡因子です。これはシーケンスデータの質を決定します。シーケンスエラー率は、シーケンスサイクルと大きく関連しており、イルミナのハイスループットシーケンスプラットフォームの一般的な特徴である試薬の消費により、各リードの終わりに向かって上昇します。
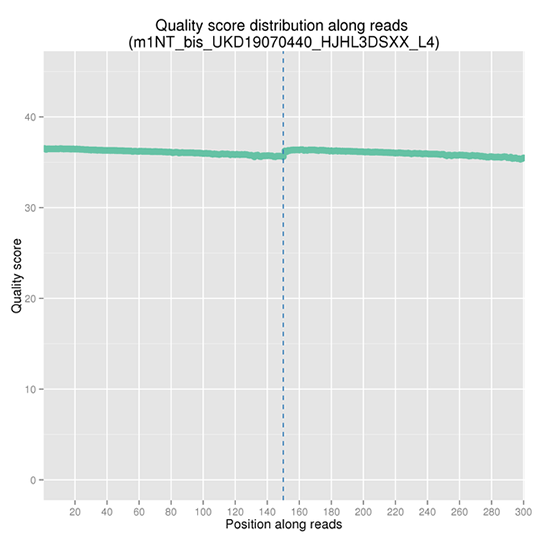
注:X軸はリードの位置を表し、Y軸はその位置における全リードの平均塩基エラー率を示します。
GC含量分布
GC含量分布は、AT/GC分離の可能性をチェックすることを目的としています。サンプルのコンタミネーション、シーケンスバイアス、ライブラリ調製時のエラーは、シーケンス結果に影響を与える可能性があります。
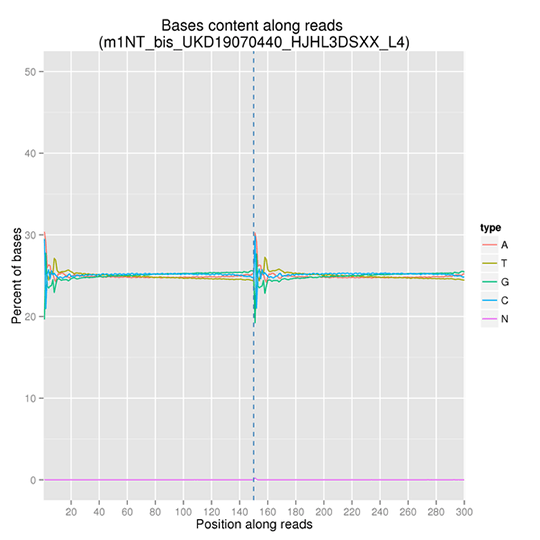
注:X軸はリード中の位置を表し、Y軸は各塩基(A, T, G, C)の割合を表します。
参照ゲノムとのアラインメントシーケンスデプスとカバレッジの分布
シーケンスデプスとカバレッジは、既知の参照ヌクレオチドにアライメントされたペアエンドクリーンリードの平均数を示しています。シーケンスカバレッジ分布は、特定の塩基位置で変異の同定がある程度の信頼性をもって行えるかどうかを決定します。
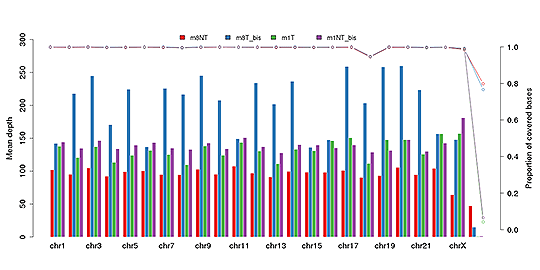
各染色体における平均シーケンスデプス(棒グラフ)とカバレッジ(点線グラフ)
注:X軸は染色体、左のY軸は平均デプス、右のY軸はカバレッジ(カバーされた塩基の割合)を示します。
SNPとInDelのコール, アノテーションと統計
一塩基多型(SNPs)は、一塩基変異体(SNVs)としても知られ、ゲノム中の遺伝的変異の最大クラスを構成しています。遺伝的変異のもう一つのクラスは、長さが50bp未満の小さな挿入や欠失(InDels)です。コーディング領域やスプライシング部位に存在するInDelは、mRNA転写産物やタンパク質に変化を引き起こす可能性があります。
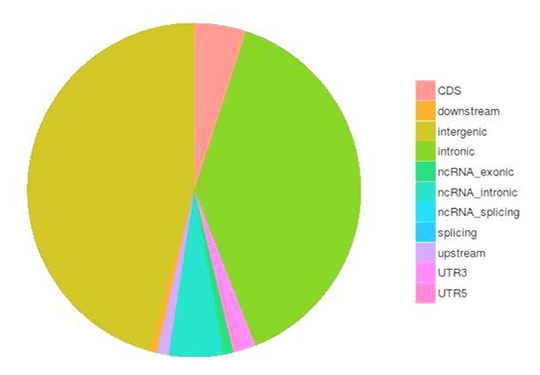
様々なゲノム領域におけるSNPs/InDelsの数
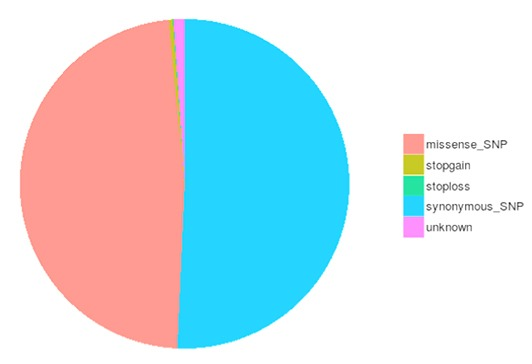
コーディング領域における異なるタイプのSNP/InDelsの数
高度な解析ドライバー遺伝子解析有意に変異した遺伝子のヒートマップ
がんに関連する変異のうち、遺伝子に影響を与えることで腫瘍形成を促進できるものはわずかである。有意に変異した遺伝子(SMGs)とは、バックグラウンド変異率(BMR)よりも有意に高い変異率を示す変異を指し、したがって腫瘍形成の間のポジティブセレクションを示します。SMGsの解析は、がんの発生と進行に重要な遺伝子を特定するのに役立ちます。
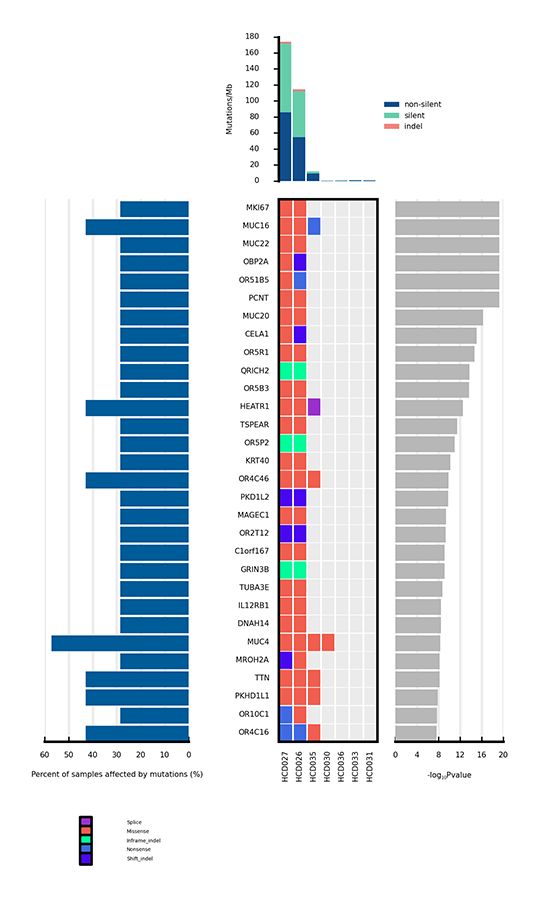
サンプル間で有意に変異した遺伝子(SMGs)のヒートマップ
注 上部の棒グラフは各サンプルの変異率(Mutation/Mb)を示します。中央のヒートマップは、サンプル間の各SMGの変異タイプを示します。横軸はサンプル、縦軸はSMGを表します。異なる変異タイプは異なる色で区別されています。ヒートマップの左側の棒グラフは、各SMGの変異によって影響を受けたサンプルの割合を示し、右側のプロットはSMGのp値を示します。
腫瘍不均一性解析腫瘍内不均一性解析
腫瘍内不均一性とは、腫瘍細胞の不均一な構成を指します。腫瘍内の不均一性とクローン構造を解読することは、治療抵抗性の理解に貢献する可能性があります。
体細胞変異の変異対立遺伝子頻度を解析することにより、腫瘍サブクローンの数と内容(サブクローン体細胞変異)を同定しました。
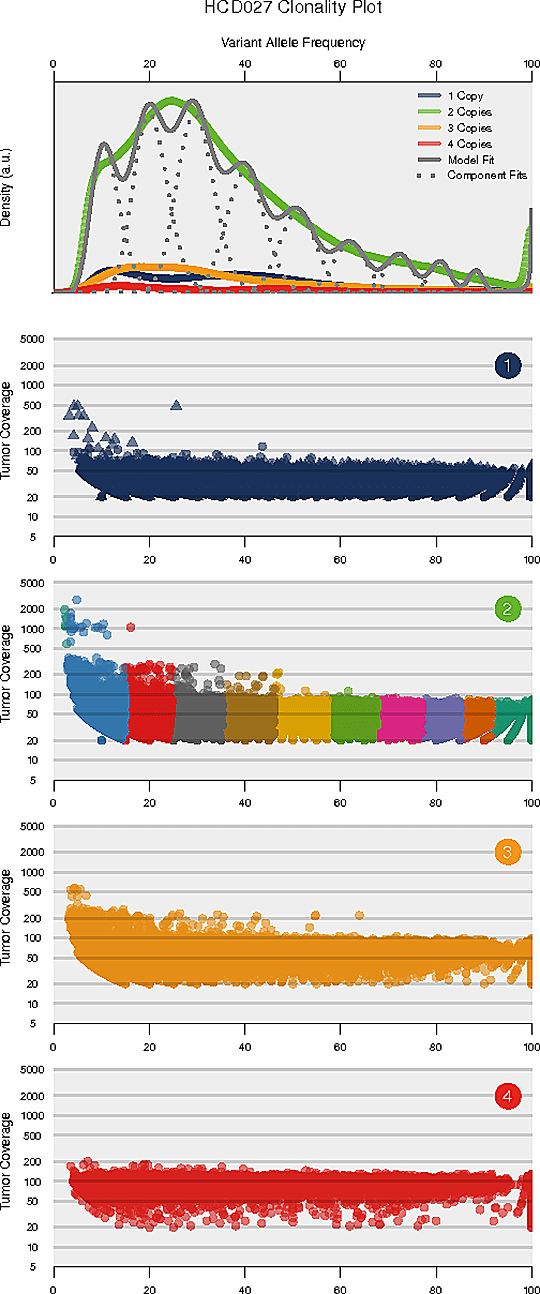
注 各パネルの横軸は変異対立遺伝子頻度(VAF)を表します。VAFが比較的低い変異のクラスターはサブクローン集団を表します。一番上のパネルは、コピー数が1、2、3の領域にわたるVAFのカーネル密度、コピー数が中立の変異体についての全クラスターを合計した事後予測密度、各クラスター/構成要素の事後密度を示しています。トップパネルの下のパネルは、コピー数領域の各クラスについて、リードデプスとVAFの関係を示しています。
*フルバージョンのデモレポートをご希望の方はお問い合わせください。