概要
ショットガンメタゲノムシーケンスは、微生物の分離、培養、標的領域の増幅の必要性を排除し、サンプル中のすべての生物から全ゲノムDNAの包括的な解析が可能です。約99%の微生物は実験室条件下では培養不可能であることを考えると、この方法は特にインパクトが大きいです。16S/18S/ITSアンプリコンシーケンスのような標的アプローチとは異なり、ショットガンメタゲノミクスは次世代シーケンス(NGS)技術を利用し、機能プロファイリング、遺伝子予測、抗菌薬耐性遺伝子の同定、コミュニティ全体における微生物相互作用のより深い理解とともに、種レベルの分類学的注釈を提供します。
広範な専門知識と最先端のバイオインフォマティクス・パイプラインを備えたノボジーンのショットガンメタゲノムシーケンスサービスは、多様なサンプルタイプの微生物のゲノム、機能、遺伝的変異を探索するためのツールを研究者に提供します。ノボジーンのソリューションは、高品質なデータ、論文発表可能な結果、幅広い研究ニーズに合わせたカスタマイズされた解析を確実にします。ノボジーンは、群集構造の特性解析や生物種の分類から、環境マイクロバイオームにおけるシステム進化、遺伝子機能、代謝ネットワークの調査まで、科学的発見を促進する包括的なメタゲノミクスソリューションを提供します。
アプリケーション
ショットガンメタゲノムシーケンスは、分類学的プロファイリング、機能解析、代謝ネットワーク研究など、幅広い研究アプリケーションに対応する汎用性の高いプラットフォームを提供します。主なアプリケーション:
- 微生物群集の包括的な解析により、分類学的および機能的プロファイルに関する洞察を得る。
- 人の健康、環境修復、持続可能なエネルギー生産に関連する微生物群の検出。
- 微生物の遺伝子設計図と代謝経路の詳細な探索による、医薬品イノベーションへの情報提供。
- 微生物とその宿主との間の複雑な関係を調査し、創薬や治療法の開発を促進する。
これらのアプリケーションは、多様な科学分野の発見を促進するショットガンメタゲノムシーケンスの有用性を強調しています。
ショットガンメタゲノムシーケンスの利点:
- ショットガンメタゲノムシーケンスでは、単離、培養、標的領域の増幅を行うことなく、複数の微生物を同時に解析できるため、原核生物や真核生物にまたがる微生物や機能の多様性に関する知見が得られます。
- メタゲノムシーケンスにおける豊富な経験を持つノボジーンは、土壌、便、水など多様なサンプルタイプに対して高品質の結果を提供します。
- ノボジーンは、アセンブリーベースの解析や、包括的なデータ解釈のための2つの異なるリードマッピング手法など、お客様の研究ニーズに合わせた幅広い解析オプションを提供しています。
仕様:DNAサンプル要件
| サンプルタイプ | サンプル量 | 液量 | 濃度 | 純度 |
| total DNA | ≥ 100 ng | ≥ 20 μL | ≥ 5 ng/μL | OD260/280 = 1.8-2.0 分解なし, 色なし, コンタミなし |
仕様:シーケンスと解析
| シーケンス プラットフォーム |
イルミナNovaSeq プラットフォーム | |
| シーケンス法 | ペアエンド 150bp | |
| 推奨シーケンスデプス |
|
|
| 解析オプション (こちらの3つからお選びください) |
標準解析 (アセンブリベース) |
|
| MetaPhlAn4-HUMAnN3 (Reads-Mapping ベース) |
|
|
| Kraken2 (Reads-Mapping ベース) |
|
|
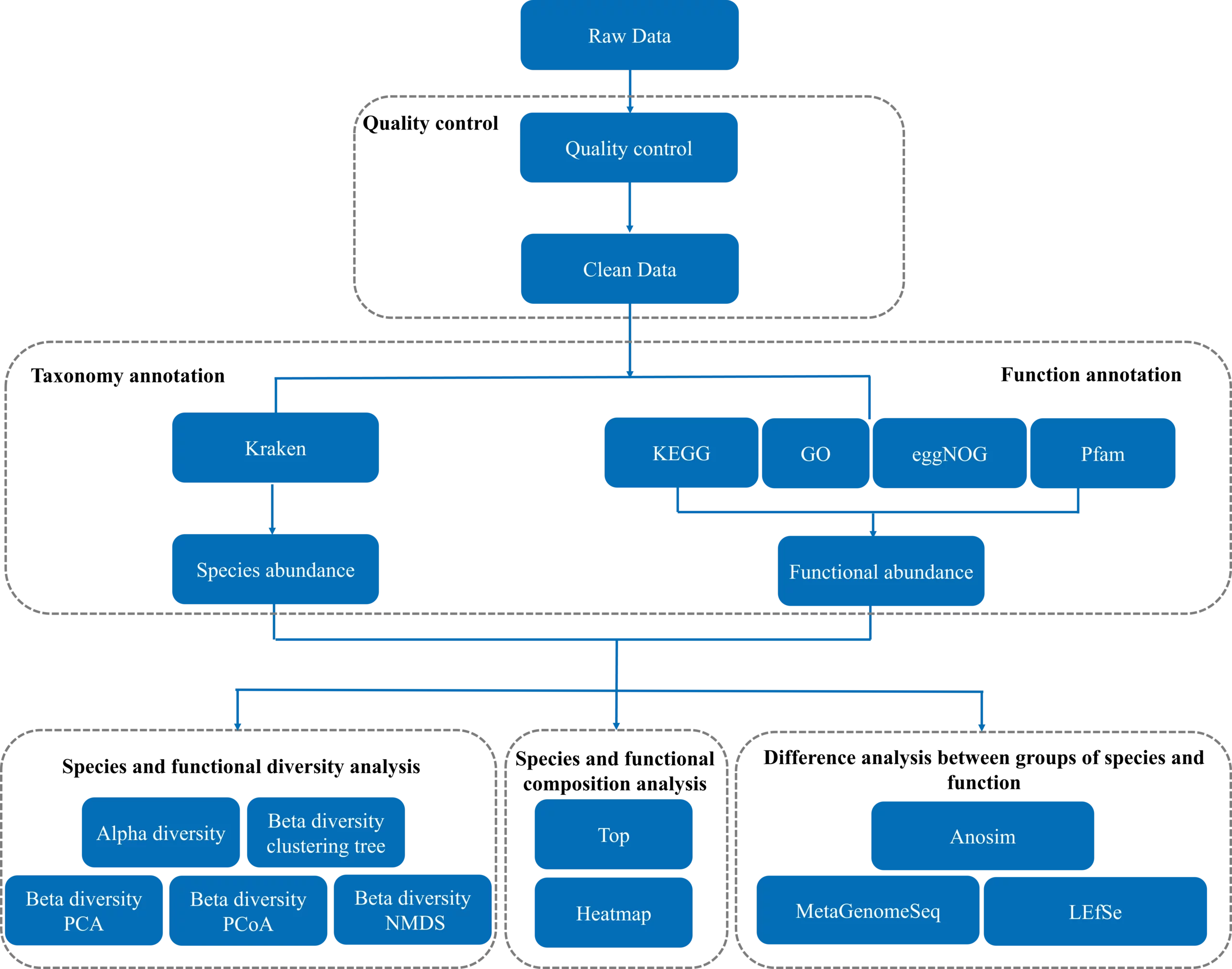
プロジェクトワークフロー




サンプル:
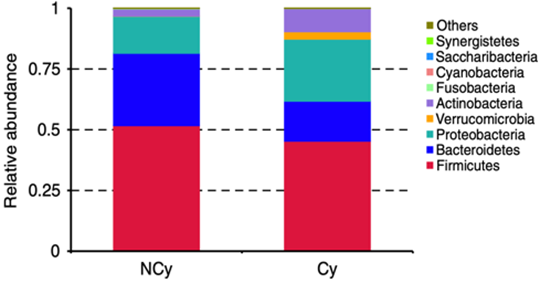
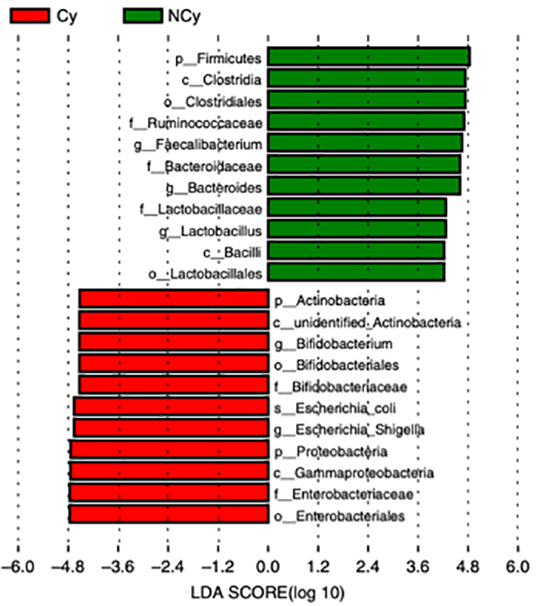
2つのグループ(NCyとCy)の先天性心疾患を持つランダムに選択された40人の子供から収集された新鮮な糞便サンプルから抽出されたゲノムDNA
シーケンス方法
Illuminaプラットフォーム、ペアエンド150 bp
結論:
CCHD患者のBMSCは、腸内細菌叢および腸内微生物叢由来のD-ガラクトースの蓄積が原因であると考えられる、早期老化の素因がありました。 Lactobacillusの補充により、欠損したBMSCが回復しました。 私たちの調査結果は、経口乳酸菌補充が自己BMSCベースの再生療法の効率を高め、CCHD患者の幹細胞ベースの治療法を改善する可能性があることを示唆しています。
Temporal Stability of the Human Skin Microbiome
緒論
地理的生物要因と個性は、人間の皮膚のマイクロバイオームの構造的および機能的構成を形作ります。 これらの要因が皮膚微生物群集の安定性に及ぼす影響を調査するために、数か月および数年にわたって収集された長期的なサンプルからメタゲノムシーケンスデータを生成しました。 複数の界に及ぶリファレンスベースのアプローチを使用して、これらのサンプルを解析したところ、皮膚が外部環境にさらされているにもかかわらず、細菌、真菌、およびウイルスの微生物叢は、時間の経過とともにほぼ安定していることがわかりました。
サンプル
12人の健康な人の皮膚から抽出されたDNAサンプル
シーケンス方法
Illuminaプラットフォーム、ペアエンド150bp

結論
サイト、個性、系統発生はすべて安定性の決定要因でした。 足のサイトが最も変動性を示した。 個人の安定性は異なっていました。 そして一過性は真核生物ウイルスに特徴的で、コロニー形成による部位特異性をほとんど示さなかった。 菌株および単一ヌクレオチドの変異レベル分析は、個人が環境から蔓延する微生物を再獲得するのではなく、維持することを示した。 皮膚微生物群集の長期間の安定性は、定着抵抗についての仮説を生成し、病状で観察された変化を探究する臨床研究に示唆を与えます。
Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life
緒論:
腸内細菌叢は人間の健康の中心ですが、初期の生命におけるその確立は、定量的かつ機能的に検討されていません。 スウェーデンの乳児とその母親の大規模コホートの糞便サンプルにメタゲノム解析を適用して、生後1年間の腸内微生物叢を特徴付け、分娩様式とその給餌の影響を評価しました。
サンプル:
DNAサンプルは、4歳(固形食品の導入時期)と12か月(一般的に子供に完全食を与えた場合)の出産時の母親と新生児を含む糞便サンプルシリーズを抽出
シーケンス方法
Illuminaプラットフォーム、ペアエンド150 bp
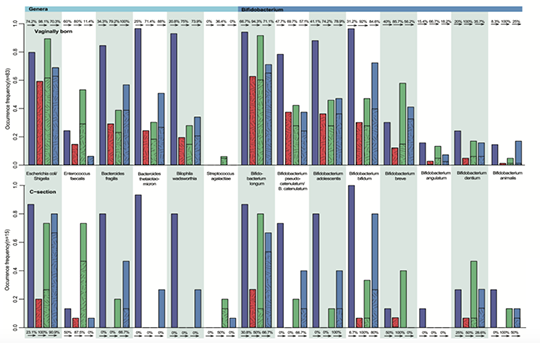
結論
おそらく、乳児の腸内で非ランダムな遷移が観察されました。恐らく嫌気性環境の確立、栄養素の利用可能性、および微生物叢の連続した相互作用によって引き起こされたと考えられます。 さらに、4,000を超える新しい微生物ゲノムの同定を可能にするMetaOTUメソッドを開発しました。これにより、さまざまな環境における微生物ゲノムの包括的な調査に非常に役立ちます。
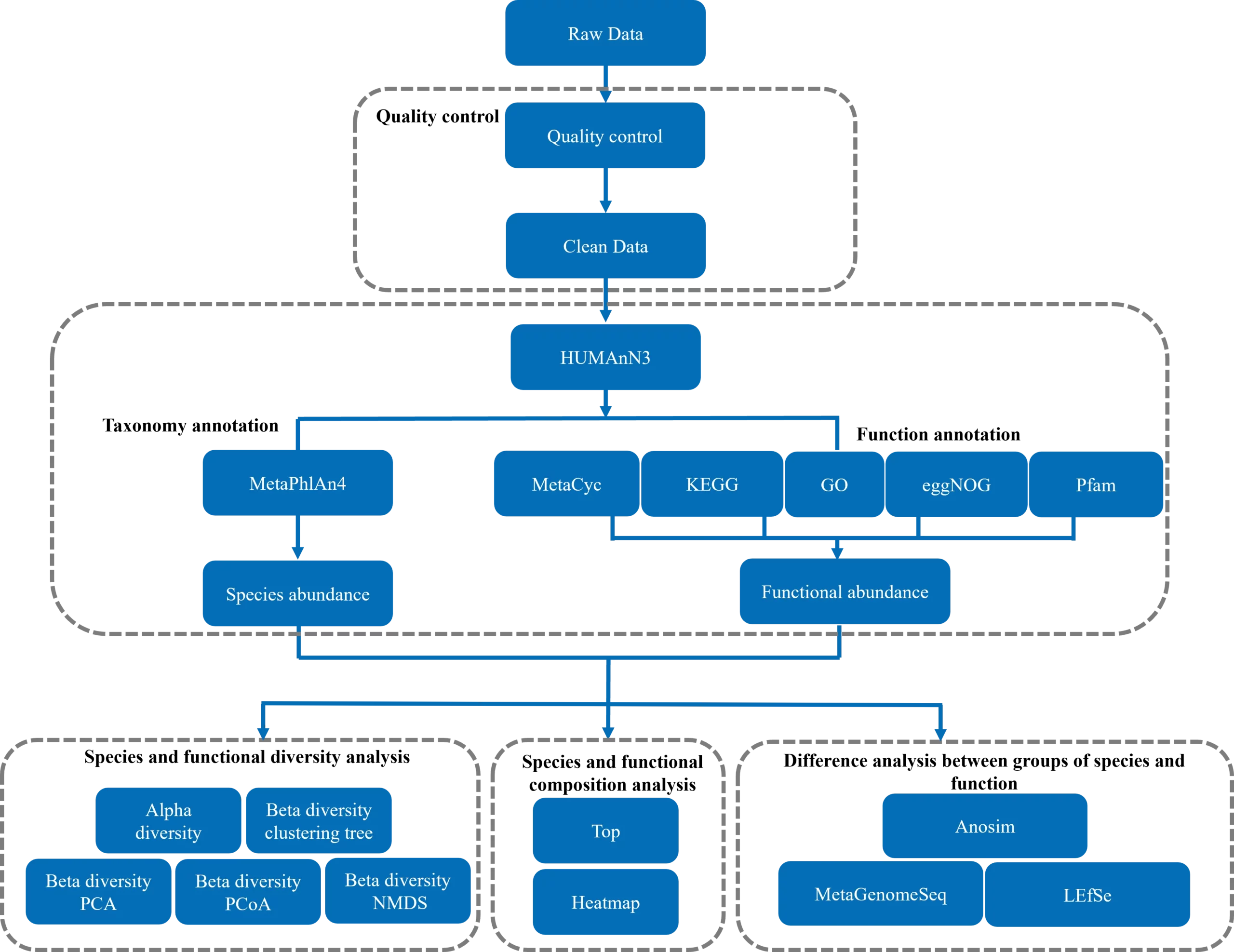
ノボジーンのアセンブリベース解析パイプライン
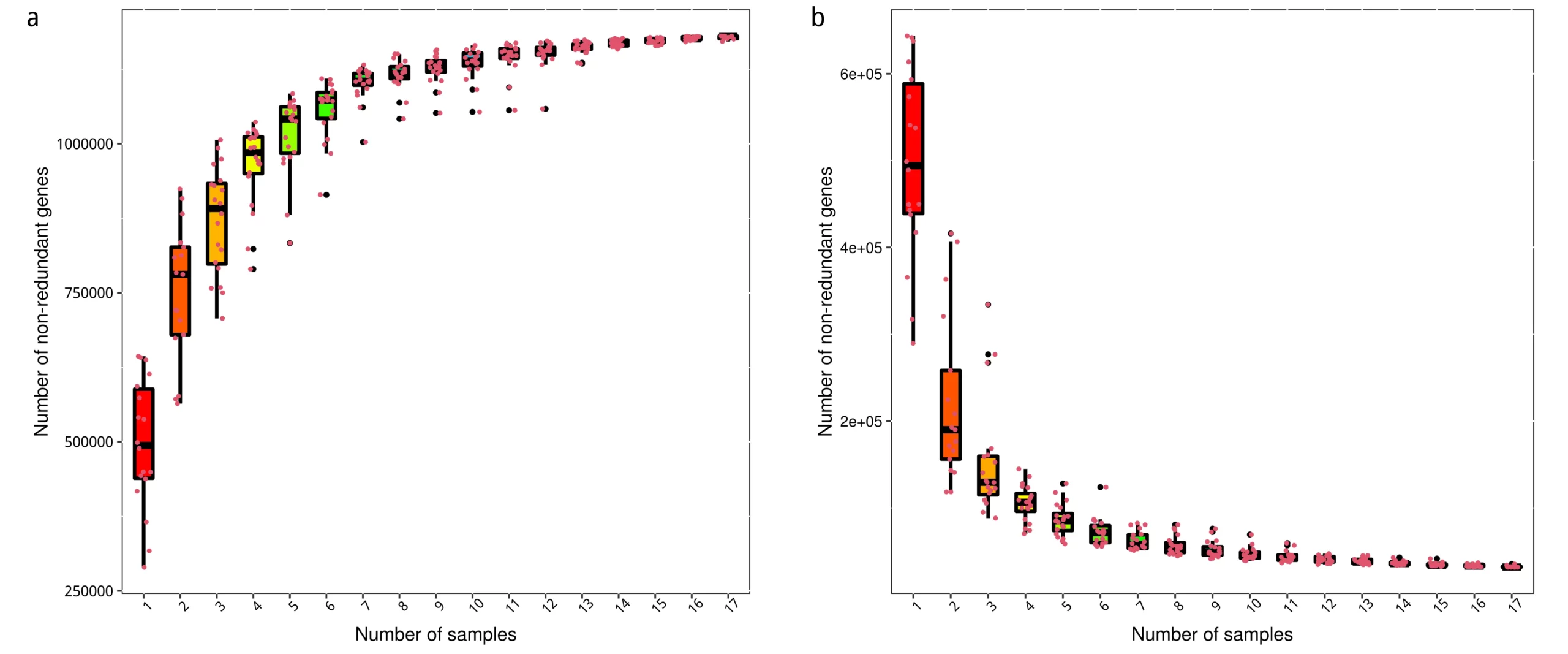
コア-パンゲノム解析
希薄化曲線は遺伝子存在量表から作成されていて、ランダムサンプリングによるコア遺伝子とパン遺伝子の数を表しています。(a) パンゲノムの希薄化曲線、(b) コアゲノムの希薄化曲線。X軸はサンプリングされたデータセット数、Y軸は遺伝子数を示します。
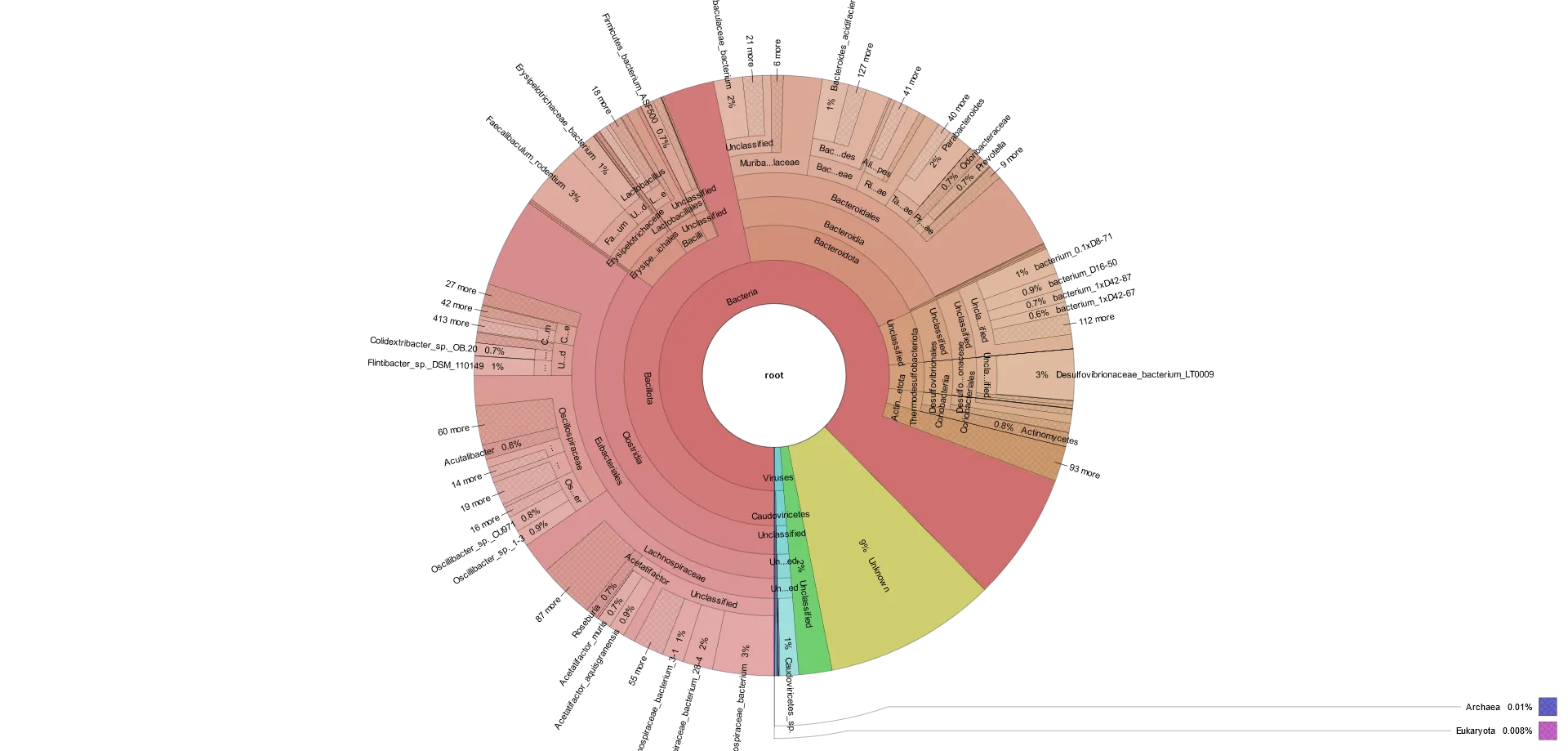
Krona可視化
Kronaは、相互作用的でズーム可能な円グラフを通して階層的データの探索を可能にし、複数の階層レベルにまたがる分類群の相対的な存在量に関する洞察を提供します。(Ondov et al., 2011) このビジュアライゼーションは、推定される存在量をより明確に表現し、分類学的分類をより深く理解することで、従来のメタゲノム解析のビジュアライゼーションを強化します。下の例では、同心円が異なる分類学的レベルを表し、セクタ領域が各分類群の存在量の割合を示しています。
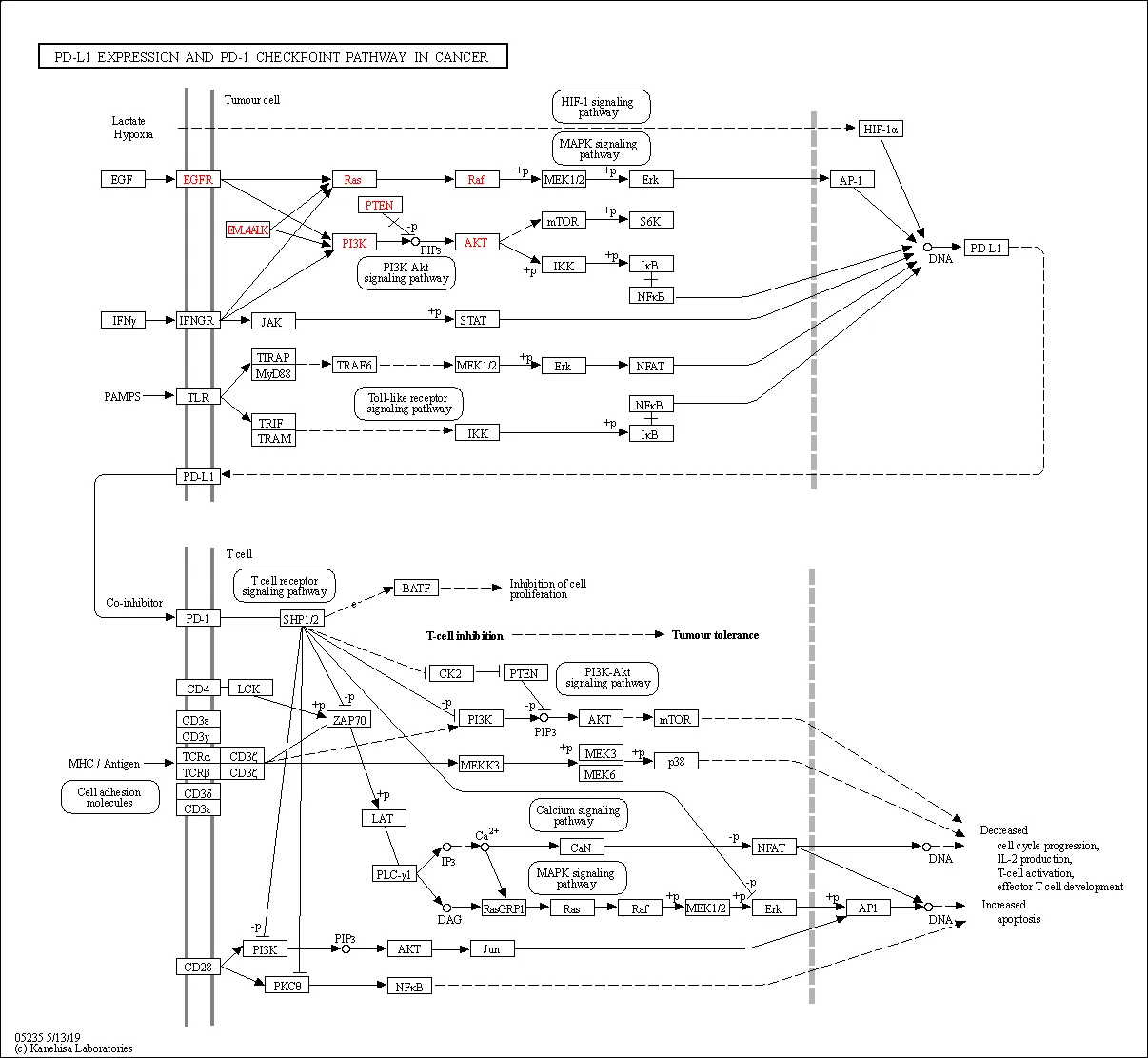
代謝パスウェイ
可視化では、ノードは化学物質を表し、長方形は酵素を表します。長方形の色の勾配(青から赤)は、一意の遺伝子数の変化を示し、より高い数は赤で表されます。色の付いた長方形にマウスカーソルを置くと、アノテーションされた遺伝子 ID が表示されます。黄色で塗りつぶされた長方形は、グループ間で大きな差がある酵素をハイライトする(該当する場合)。酵素の上にマウスカーソルを置くと、有意に変動する存在量のボックスプロットが表示されます。
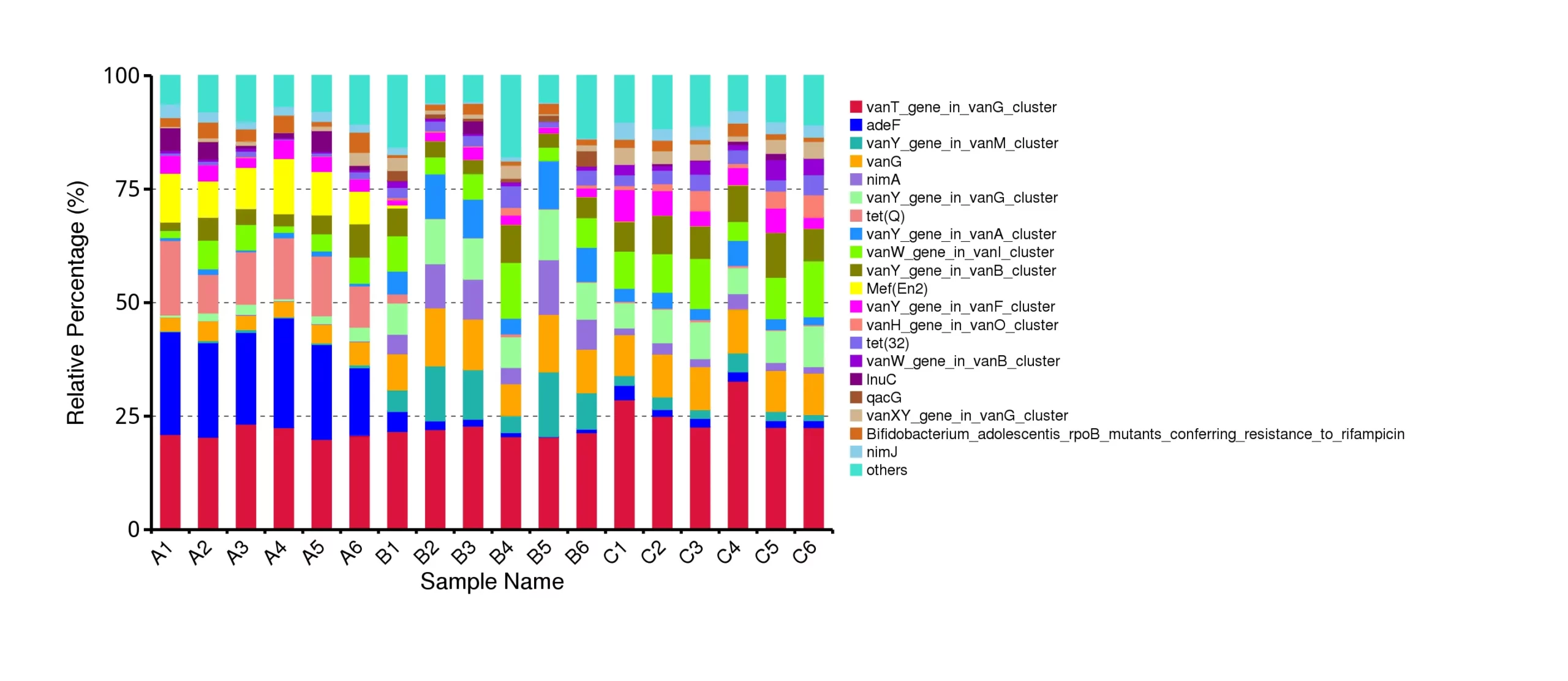
抗生物質耐性遺伝子アノテーション
結果をもとに耐性遺伝子の相対存在量を算出しています。耐性遺伝子存在量表を用いて様々な解析を行いました。凡例では、複数の耐性タイプを持つ耐性遺伝子を略号で示しています。図は、各サンプルにおける耐性遺伝子の割合分布を示しています。
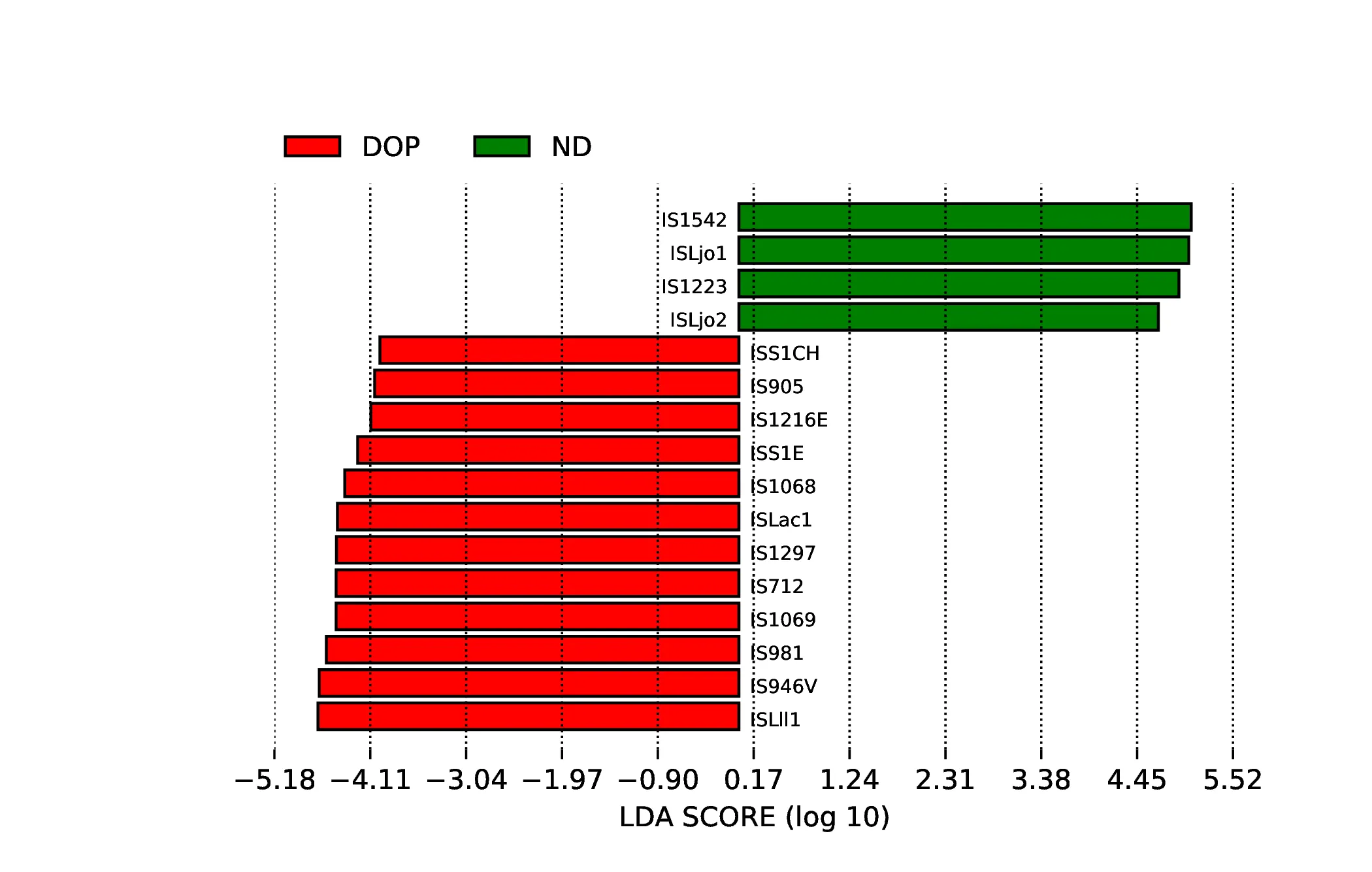
移動遺伝要素LEfSe解析
インテグロンのLDAスコアヒストグラムは、LDAスコアがデフォルトの閾値である3を超えるバイオマーカーをハイライトしています。LDAスコアは、グループ間の表現型の違いを説明するバイオマーカーの能力を反映する効果量を示します。
ノボジーンのMetaPhlAn4-HUMAnN3解析パイプライン
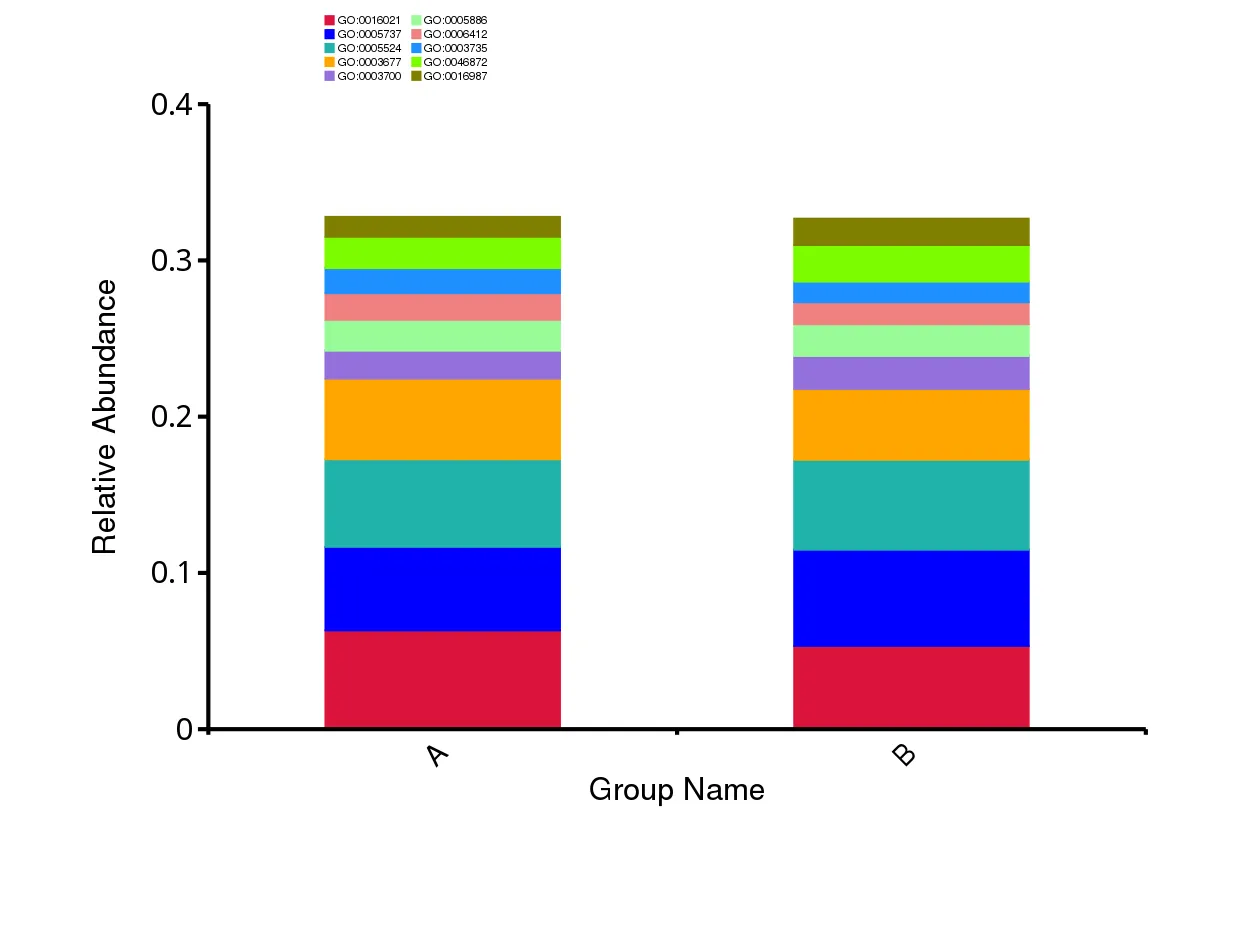
分類学的注釈の相対的存在量
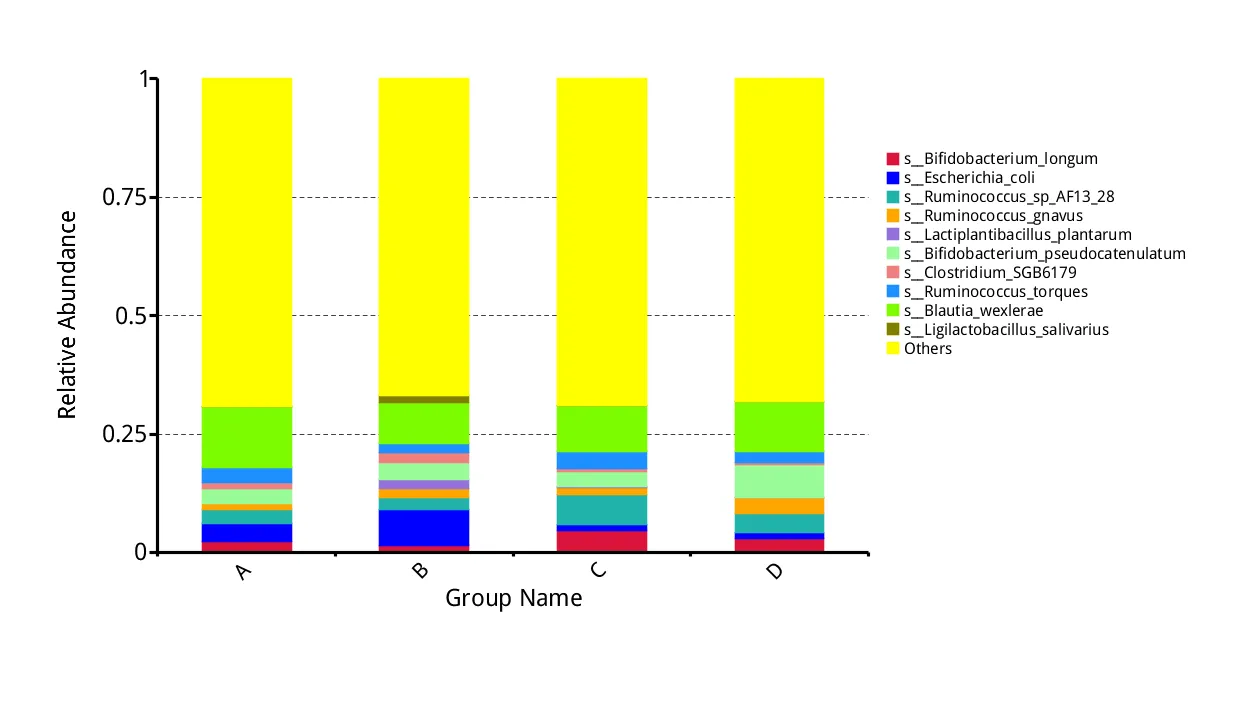
分類学的存在量表から上位10分類群(種)を選択しています。棒グラフは各サンプルまたはグループの相対的な分類学的存在量を示しています。横軸はサンプル名またはグループ名、縦軸は相対的な存在量の比率を示しています。各色ブロックは、凡例に示すように、特定の分類学的レベルに対応しています。
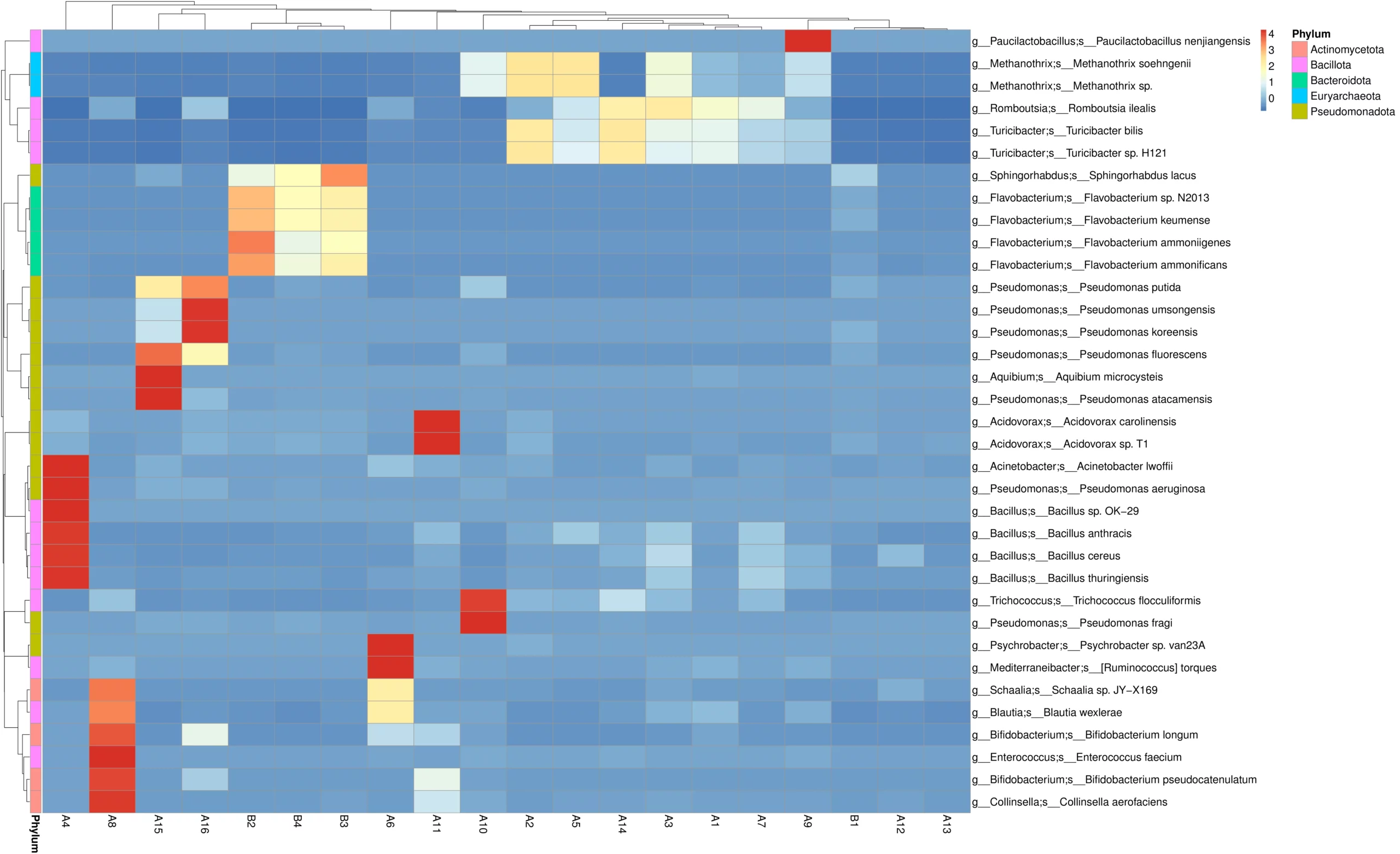
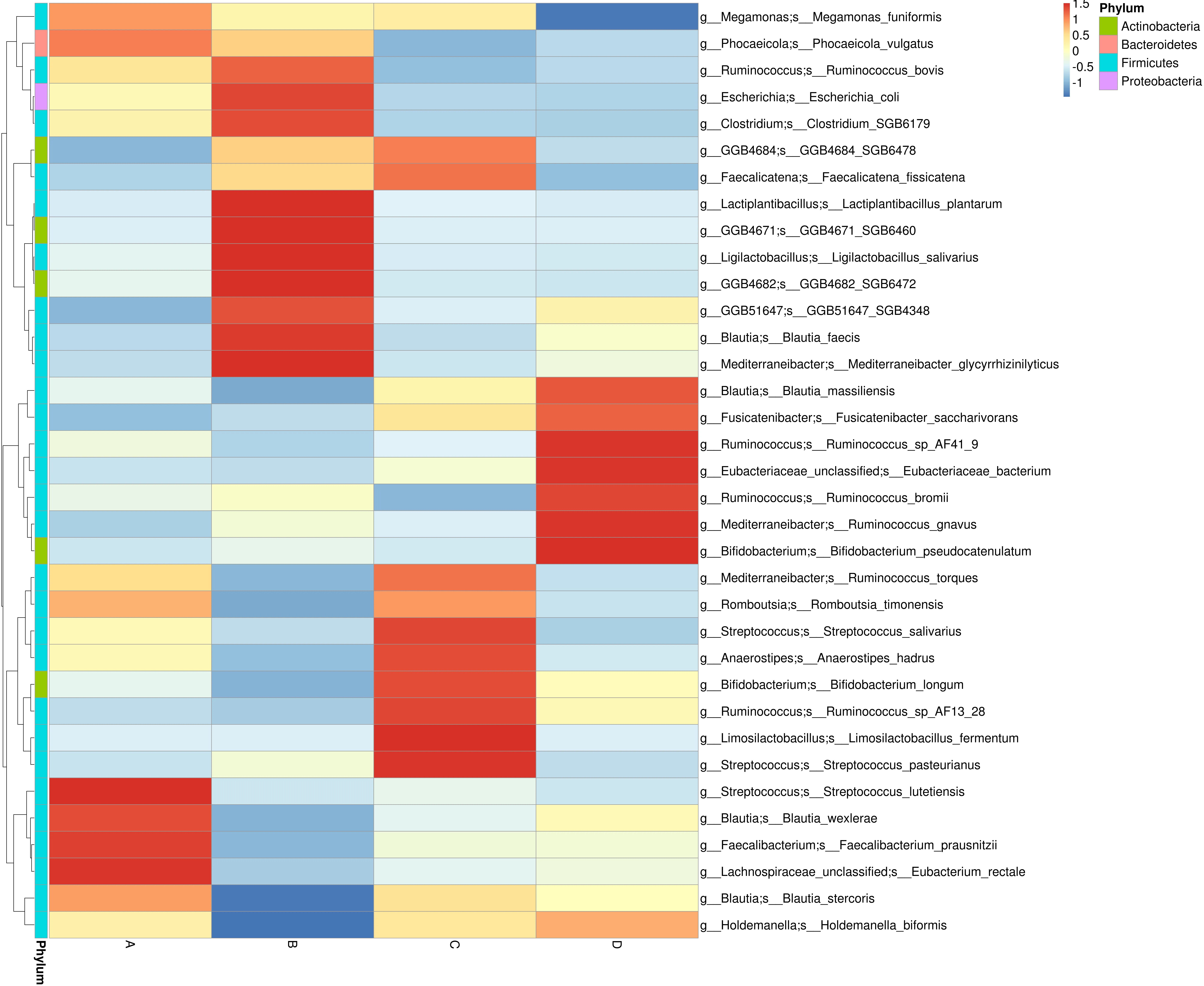
MetaPhlAn分類学的存在量ヒートマップ
ヒートマップは、全サンプルにわたる上位35の支配的な分類群(種)の存在量分布を表示します。X軸はサンプル名またはグループ名、Y軸は分類学的情報を示します。左側にはクラスタリングツリーが表示されます。ヒートマップ上の値はZスコアで、各分類群の相対存在量から正規化されている。右側の色の凡例は、ヒートマップの色とZ値の相関を示し、負のZスコアは平均値より低い値を、正のZスコアは平均値より高い値を示します。
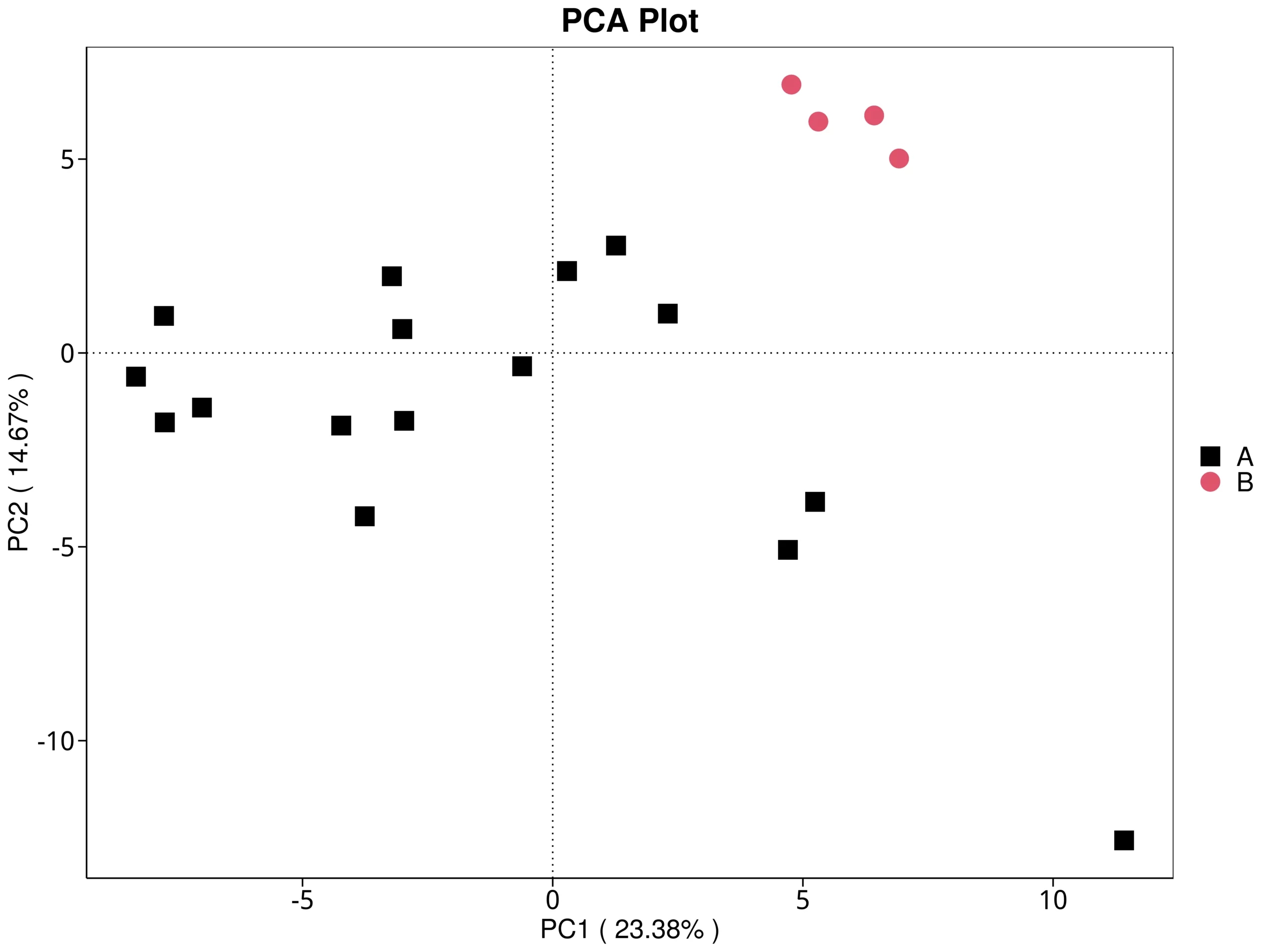
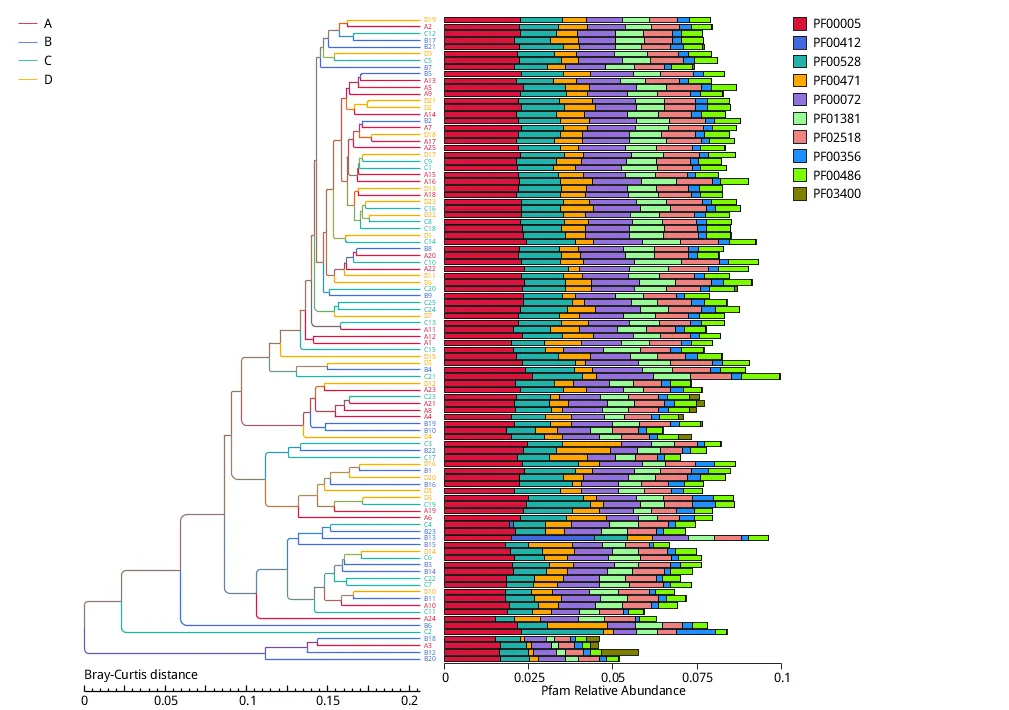
Pfamクラスター解析
Bray-Curtis距離に基づくサンプルのクラスタリングは、相対的な分類学的存在量を用いてサンプルの類似性を評価します。タンパク質ファミリーの存在量表から得られたクラスタリング結果を以下に示します。この図は、左側にBray-Curtis距離のクラスタリングツリーを、右側にタンパク質ファミリー間の各サンプルの相対存在量分布を示したものです。
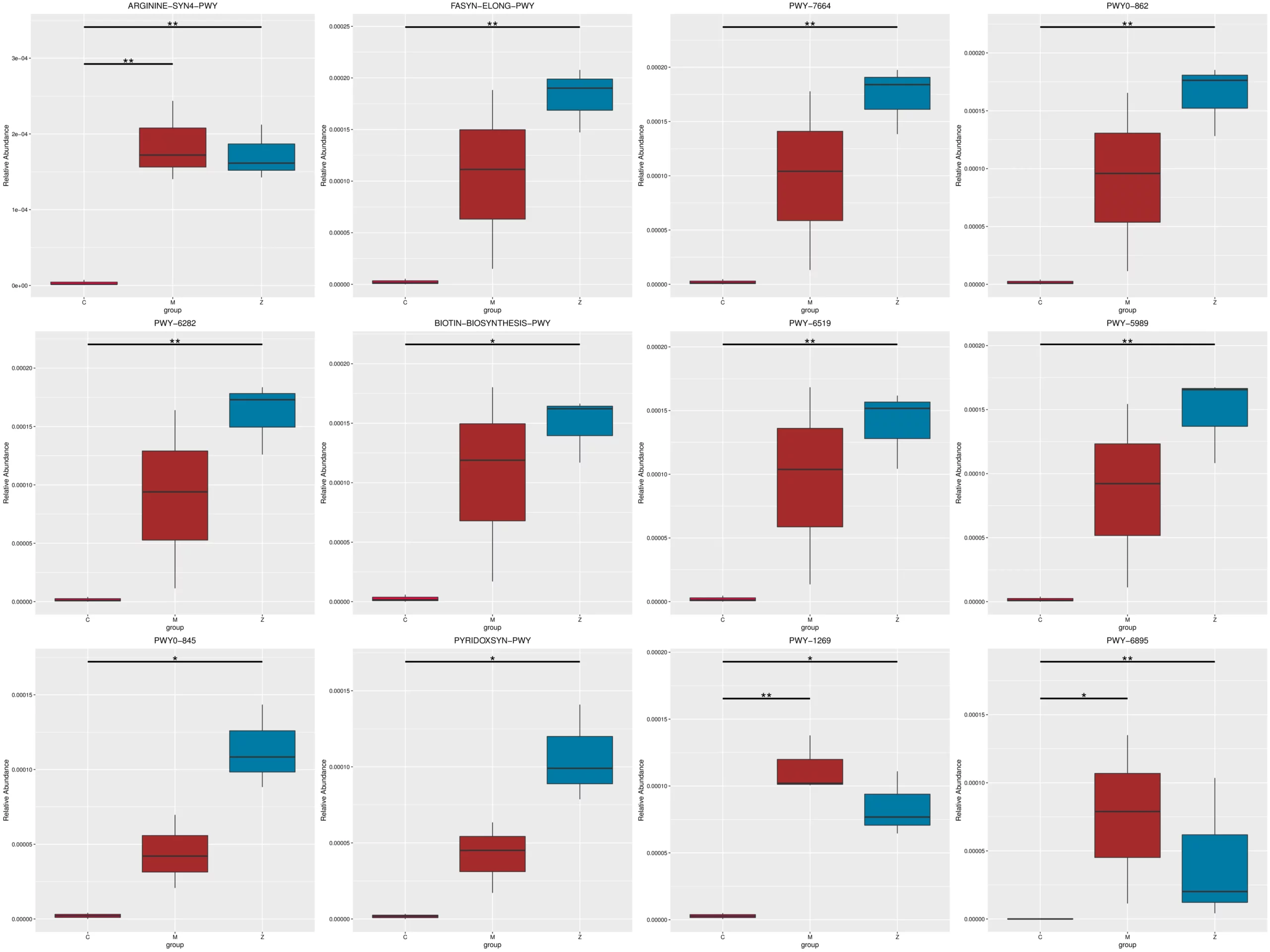
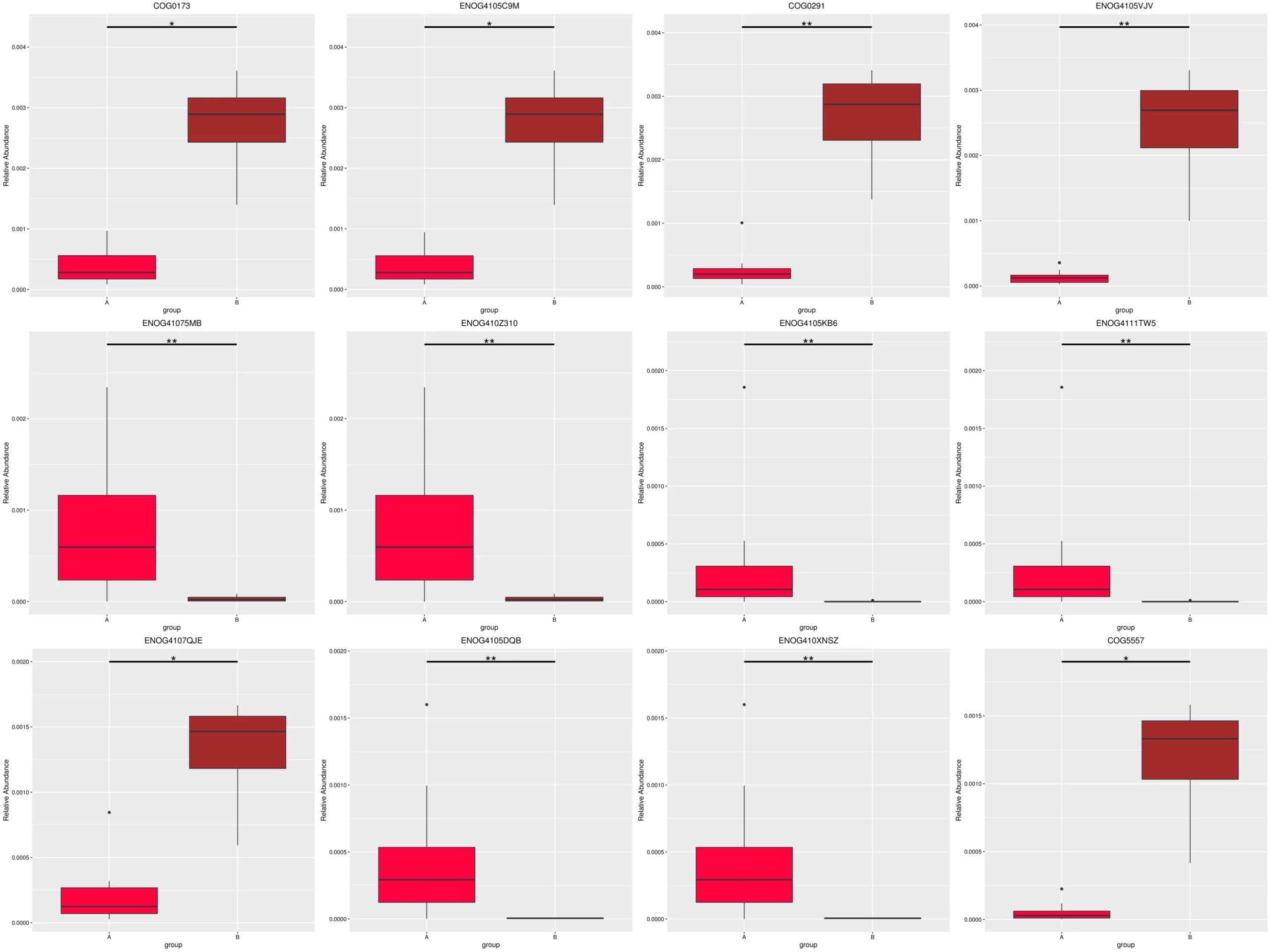
MetaCycMetaGenomeSeq
MetaGenomeSeq (Paulson et al., 2013)は、ノンパラメトリックt検定を用いて複数サンプルのメタゲノムデータを解析し、異なる存在量の特徴を検出します。p-値はq-値に調整され、有意差のある分類群を同定します。代謝経路の存在量表に基づく結果は、Y軸に存在量値を示し、サンプルグループの比較を表示します。“*”は有意な変動(q < 0.05)を示し、 “**”は極めて有意な変動(q < 0.01)を示します。