RNAシーケンス(RNA-seq)は、細胞機能の研究に変革をもたらし、研究者に細胞の転写ランドスケープに対する前例のない洞察を提供しています。ハイスループットで正確な次世代シーケンサー技術(NGS)を採用することで、RNA-seqは遺伝子発現 プロファイルを明らかにし、トランスクリプトームの連続的な変化を説明することができます。RNA-seq技術では、一本鎖のメッセンジャーRNA(mRNA) を選択的にキャプチャーまたは濃縮し、ライブラリー調製のために相補的DNA(cDNA)(mRNAs) に変換します。
ノボジーンでは、ペアエンド150bpシーケンス戦略(ショートリード)を利用する最先端のイルミナNovaSeqプラットフォームを使ってcDNAライブラリをシーケンスします。豊富な経験と強力なシーケンス能力とともに、ノボジーンは様々な研究目的に対応するサービスを提供しています。真核生物のmRNAシーケンス(mRNA-seq)サービスだけでなく、原核生物の転写産物、ノンコーディングRNA、完全長アイソフォーム(ロングリード)、全トランスクリプトーム、メタトランスクリプトームに関するデータも提供することが可能です。
RNAシーケンスのアプリケーション
mRNA-seqは、細胞のトランスクリプトームプロファイルを解析するための強力なツールです。ノボジーンのプロフェッショナルサービスは、以下のような幅広いアプリケーションで研究目標をサポートします:
- 様々な条件や処理のもとでの異なる組織やサンプルの転写物の定量的プロファイリング
- 新規転写物、選択的スプライシング(AS)、 転写物の変異の発見
- 組織特異的な転写物や時間経過に伴う遺伝子発現を利用した発生メカニズムや薬剤耐性に関する研究
- 新規転写産物/アイソフォーム、SNP/InDel同定、融合遺伝子解析に基づくバイオマーカー探索
- トランスクリプトームと組み合わせたオミックス解析
- 臨床診断における発症メカニズムやクリニカルサブタイプの検討
ノボジーンRNAシーケンスの利点
- ノボジーンのmRNA-seqは、ハイスループットと高い精度 (Q30スコア≥85%))を提供し、必要な初期RNA投入量も少なくて済みます。RNA-Seqサービスを提供する豊富な経験を有し、何千ものプロジェクトを成功させ、多くの研究者がインパクトファクターの高いジャーナルで発表することを支援しています。
- ノボジーンは、定量化、 遺伝子発現の差異、 新規転写物のアノテーション、 代替スプライシング、 融合遺伝子の発見、その他の潜在的な変化に対する包括的なソリューションを提供します。高度な資格を持つバイオインフォマティシャンが、参照ゲノムがある種、ない種を問わず、パーソナライズされたパイプラインを使用して、論文投稿可能なデータを提供します。
RNA-seq仕様:サンプル要件
| Library Type | Sample Type | Amount | RNA Integrity Number (Agilent 2100) | Purity (NanoDrop) |
| Eukaryotic RNA-Seq (cDNA library) | Total RNA | ≥ 200 ng | ≥ 4.0, with smooth base line | A260/280 = 1.8-2.2 A260/230 ≥ 1.8 |
| Total RNA (Blood) | ≥ 400 ng | ≥ 5.8, with smooth base line | ||
| Amplified cDNA (double-stranded) | ≥ 100 ng | Fragments between 400bp and 5000bp with main peak at ~2000bp | A260/280 = 1.8-2.0 A260/230 ≥ 1.8 |
|
| Eukaryotic RNA-Seq (strand specific library) | Total RNA | ≥ 400 ng | ≥ 5.8, with smooth base line | A260/280 = 1.8-2.2 A260/230 ≥ 1.8 |
注:サンプル量は参考として記載しています。詳しくは、「 サービス仕様書」または「サンプル要件 」をダウンロードしてご確認ください。詳細な情報については、「カスタマイズされたリクエストについて」とご連絡ください。
RNA-seq仕様:シーケンスと解析
| Sequencing Platform | Illumina NovaSeq 6000 Sequencing System |
| Read Length | Paired-end 150 bp |
| Data Output |
|
| Data Analysis Capability |
|
注:表示される推奨データ出力や解析内容は、あくまで参考です。詳しくは、 サービス仕様書 をダウンロードしてください。詳細な情報については、「カスタマイズされたリクエストについて 」とご連絡ください。
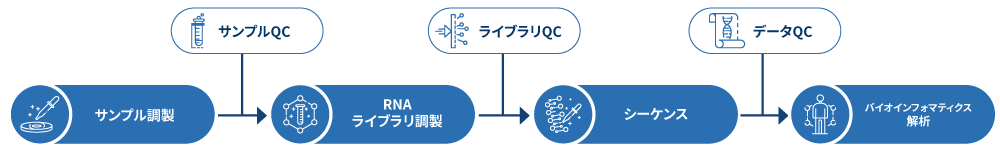
RNA-seqサービスのプロジェクトワークフロー
プロジェクトのワークフローは、お客様のサンプルがRNA-Seqの基準を満たすことを確認するためのサンプル品質管理(Sample QC)から始まります。それから、ターゲットとなる生物とアプリケーションに応じて適切なライブラリを作製し、その品質をテストします( (ライブラリQC)。次に、150bpのペアエンド シーケンスを用いてサンプルのシーケンスを行い、得られたデータの品質もチェックします(Data QC)。最後に、バイオインフォマティクス解析を行い、論文掲載可能な結果をご提供いたします。以下のフローシートは、mRNA-seq技術フローの段階的なプロトコルを説明したものです。
サンプルの調製に続き、RNAライブラリの調製を行います。RNAライブラリーは、polyAキャプチャー(またはrRNA除去法)およびcDNAの逆転写によって形成されます。イルミナPE150テクノロジーでサンプルのシーケンスを行い、最終段階でバイオインフォマティクス解析を行います。




FAQS
ノボジーンのRNA-seqサービスを利用した注目の論文
RNA-seq(mRNA-seq)は、最も頻繁に引用されるNGSメソッドです。ここでは、ノボジンのRNAシーケンス(mRNAシーケンス)サービスを利用した優れた学術論文をまとめています。
-
Cell Death & DifferentiationIssue Date: 2021.2IF: 10.717DOI: 10.1038/s41418-021-00749-4
-
Stem Cells Translational MedicineIssue Date: 2021.1IF: 11.5DOI: 10.1002/sctm.20-0468
-
Journal of Hazardous MaterialsIssue Date: 2020.12IF: 9.038DOI: 10.1016/j.jhazmat.2020.124867
-
Genome MedicineIssue Date: 2020.11IF: 10.675DOI: 10.1186/s13073-020-00796-5
-
Advanced scienceIssue Date: 2020.3IF: 15.84DOI: 10.1002/advs.202000398
Error Rate Distribution
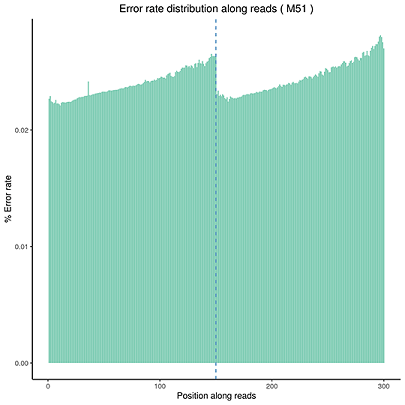
The x-axis shows the base position along each sequencing read and the y-axis shows the base error rate.
GC Content Distribution
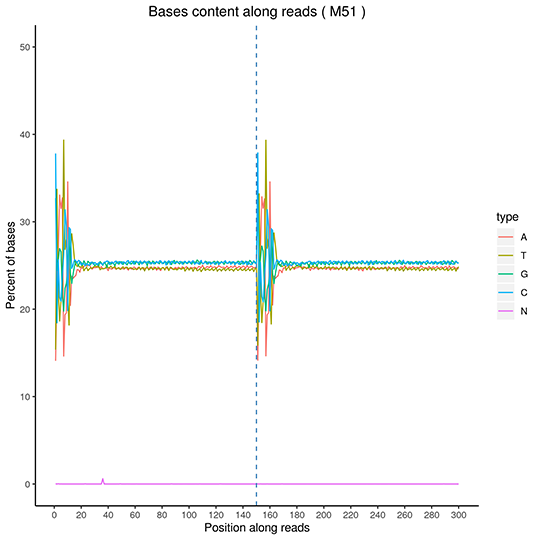
The x-axis for reads position, the y-axis for single base percentage. Different color for different base type.
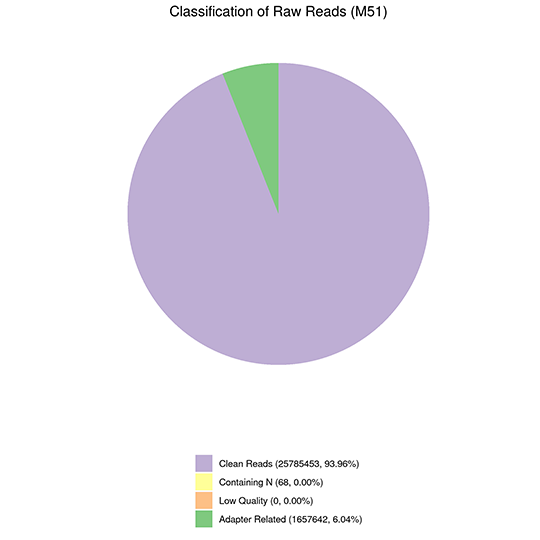
Classification of Raw Reads
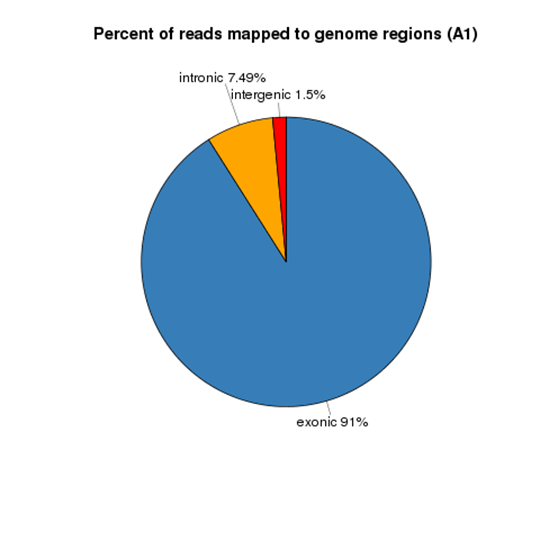
Reads Distribution on Reference Genome
Gene Expression Quantification
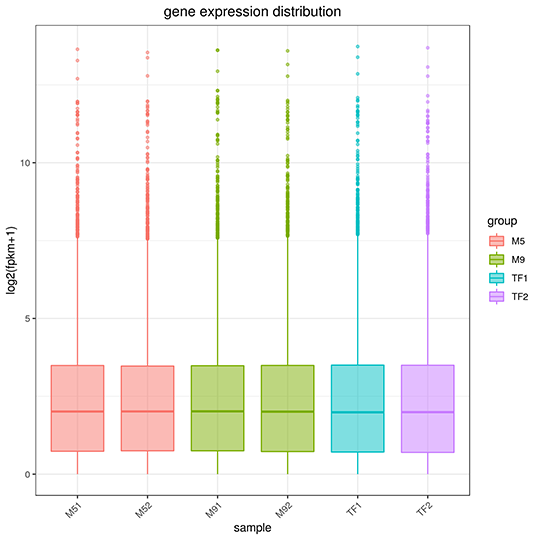
The x-axis represents the name of sample, the y-axis indicates the log10(FPKM+1), parameters of box plots are indicated, including maximum, upper quartile, mid-value, lower quartile and minimum.
Volcano Plot of changes on Gene Expression
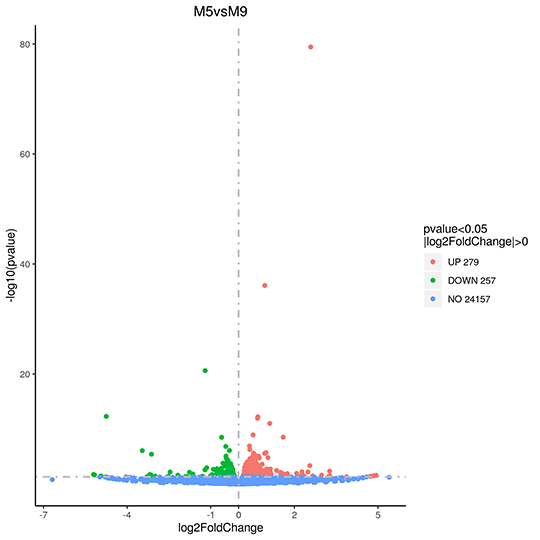
The x-axis shows the fold change of genes in different samples. The y-axis shows the statistically significant degree of changes in gene expression levels. The smaller the corrected pvalue, the bigger -log10(corrected pvalue), the more significant the difference. The points represent genes, blue dots indicate no significant difference in gene expression, red dots indicate upregulated differentially expressed genes, green dots indicate downregulated differentially expressed genes.
Hierarchical Clustering Heatmap of Differential Expression
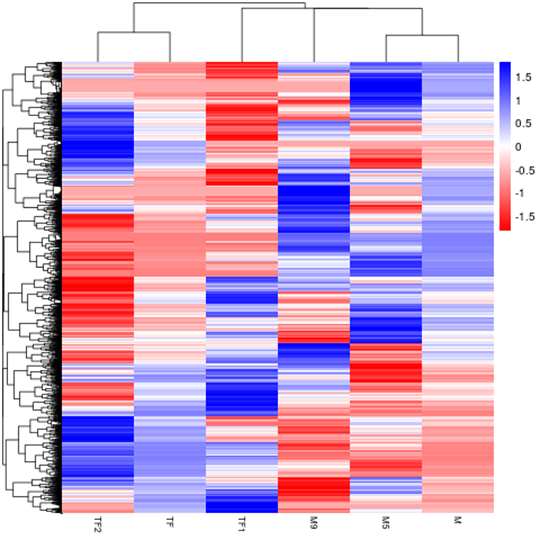
The overall results of FPKM cluster analysis, clustered using the log10(FPKM+1) value. Red denotes genes with high expression levels, and blue denotes genes with low expression levels. The color ranging from red to blue indicates log10(FPKM+1) value from large to small.